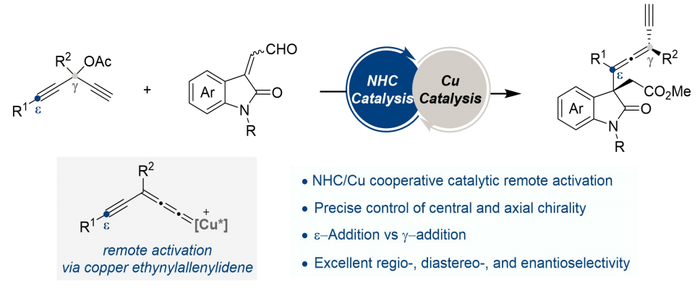
【最新科研】铜和手性氮杂环卡宾协同催化远程活化实现中心手性和联烯轴手性的精准
(图片来源:Angew. Chem. Int. Ed.)

正文

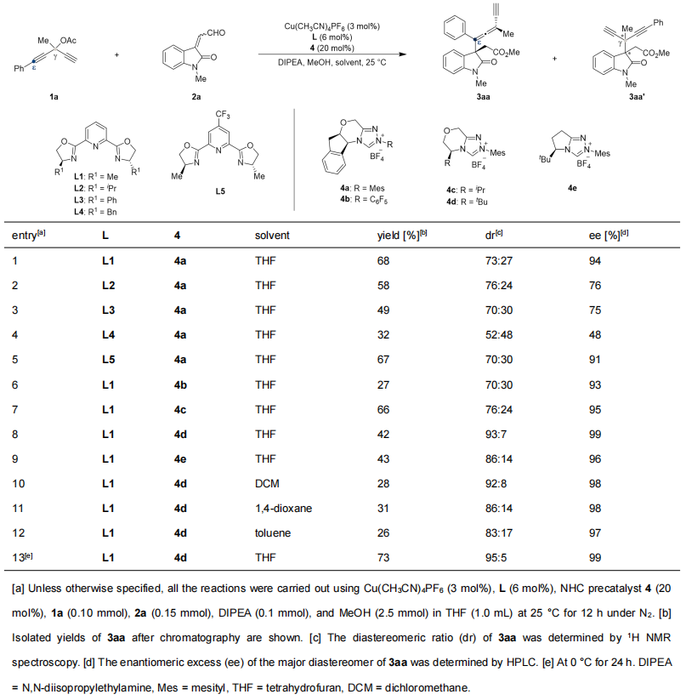
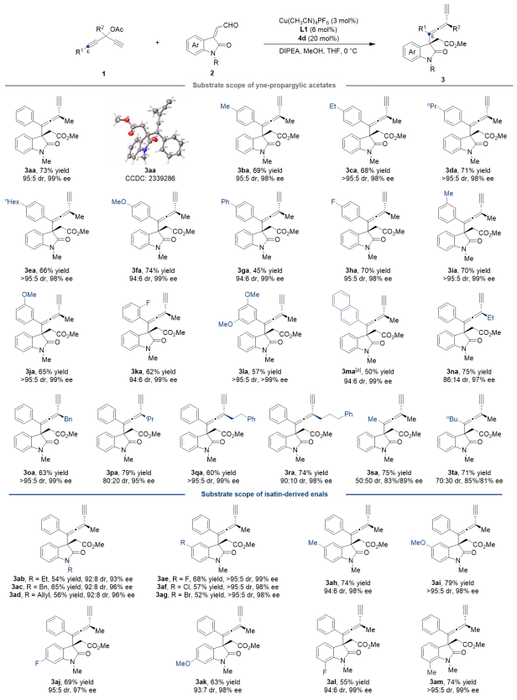
作者以苯乙炔基取代的炔丙基醇酯1a和氧化吲哚衍生的α,β-不饱和醛2a为原料进行反应尝试 (表1)。使用甲基取代的手性吡啶双噁唑啉配体L1和茚醇衍生的手性氮杂环卡宾4a,四氢呋喃为溶剂,甲醇为淬灭试剂,二异丙基乙基胺作碱,在室温下反应12小时,可以以68%的收率、73:27 dr的非对映选择性以及94% ee的对映选择性获得目标产物 (表1, entry 1)。随后,作者对其他反应条件进行了筛选,确定了反应的最优条件,能够以73%的收率、95:5 dr的非对映选择性和99% ee的对映选择性得到目标产物3aa (表1, entry 13)。
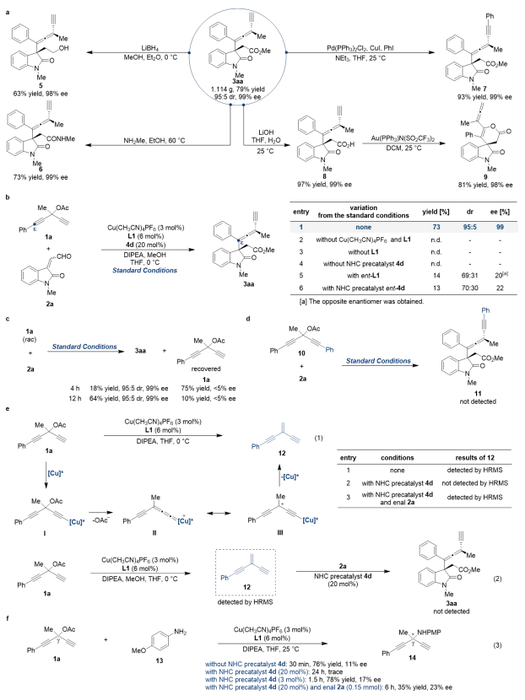
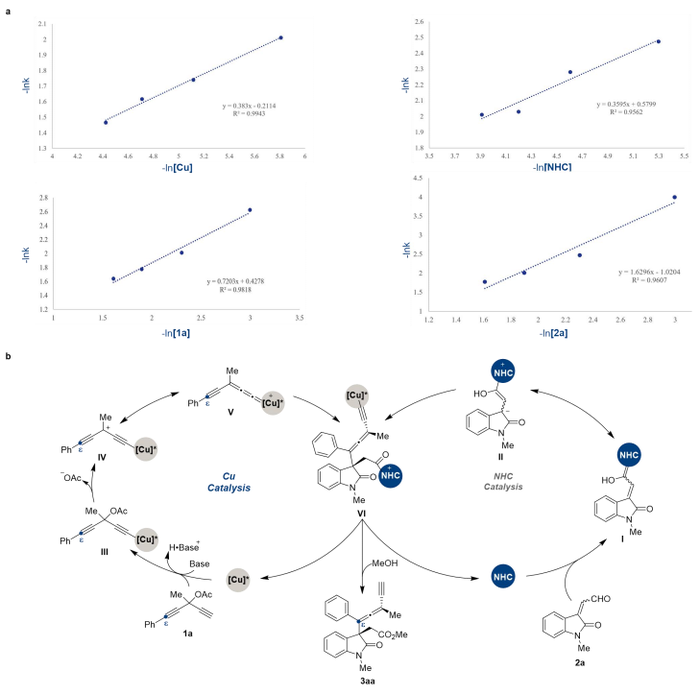
进一步的动力学实验结果表明该反应的速率与手性铜配合物催化剂、手性氮杂环卡宾催化剂、炔丙醇酯和烯醛均为一级动力学相关 (图3a)。基于实验结果和文献报道,作者提出了可能的反应机理 (图3b):手性氮杂环卡宾催化剂与烯醛2a反应生成高烯醇中间体Ⅰ;手性铜配合物催化剂与化合物1a反应,在碱的作用下生成炔基铜中间体Ⅲ,随后离去乙酸根负离子生成炔基联烯铜中间体Ⅴ,随后与高烯醇盐中间体Ⅱ反应生成中间体Ⅵ,最后醇解得到目标产物3aa并循环催化剂。

总结

化学加
展源
何发
热点文章
-

一文看懂测量不确定度
2024-09-04
-

分光光度法基本原理及应用
2024-08-05
-

各种缓冲溶液的配制方法大全
2024-10-15
-

GB/T 5750-2023《生活饮用水标准检验方法》系列标准
2024-09-02
-

MTT实验IC50,GI50,EC50还傻傻分不清楚?
2024-08-06
-

【收藏】六个快速查找抗体的神仙网站
2024-08-14
-

【小技巧】说说抽滤那些事儿
2024-09-14
-

洞察未来,共谋发展 “ 数·智·未来 ” 安捷伦未来实验室媒体圆桌会成功举办
实验室是科技创新的基础条件和成果产出源泉。十四五以来,国家着力打造战略科技力量,推进国家实验室建设和国家重点实验室体系重组,数字化、智能化、自动化赋能生物科技快速发展,掀起了科研领域创新变革的浪潮。
作者:
-
食品检验理化常用国家标准与要点
-
水分测定方法开发研究&检测相关问题故障分析解决解读
-
药物常用的晶型表征方法

评论
加载更多