研发人必备技术:TLC薄层层析色谱法
薄层色谱分析法,是利用各成分对同一吸附剂(如硅胶)吸附能力不同,使得流动相(溶剂)流过吸附剂过程中,连续的产生吸附、解吸附、再吸附、再解吸附,从而达到各成分互相分离的目的。关于薄层色谱的知识现在就跟小析姐一起来看看吧。
薄层色谱,或称薄层层析(thin-layer chromatography),是以涂布于支持板上的支持物作为固定相,以合适的溶剂为流动相,对混合样品进行定性与定量分析、分离和鉴定的一种层析分离技术。20世纪50年代从经典柱色谱法及纸色谱法的基础上发展起来的一种平面色谱技术;至20世纪60年代后,人们对薄层色谱法在使用器材的规格、操作方法及术语使用的标准化等方面进行了大量的工作,使该方法日趋成熟和完善,广泛地应用于有机合成中。TLC薄层层析技术作为有机合成中,最直接的监测反应的手段,本文对其内容进行简单的介绍。
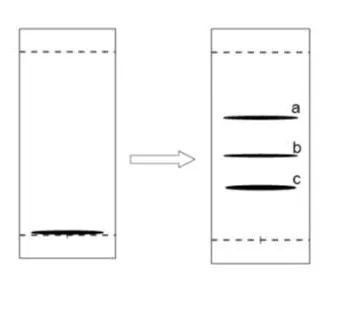
Prep-TLC分离样品:具体请看往期文章【制备薄层色谱(爬大板)纯化操作流程】
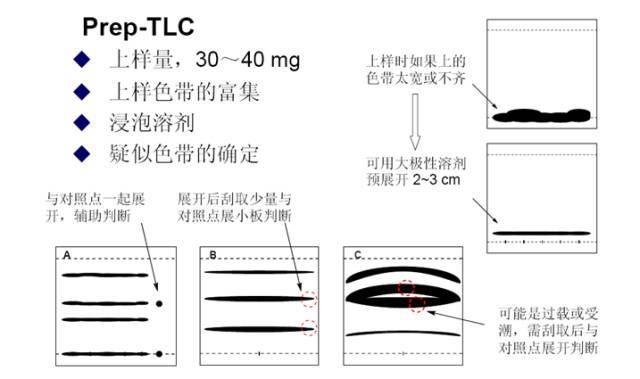
2、样品对硅胶的吸附能力过强导致的拖尾。对不同体系加入不同的调节剂,酸体系加冰醋酸或甲酸,碱体系加氨水、三乙胺或二乙胺。
3、展开剂的极性与样品极性不附,不能做到有效展开导致。可以通过调节展开剂极性解决。
4、如果是长带状,那最可能的原因是展开剂对样品的溶解度不够所导致。可以根据极性表换极性相近的对样品溶解度更好的溶剂做展开剂,另外样品未溶解完全,点在班上的样品有未溶固体样品也会导致长条状拖尾。
展源
何发
相关文章
-

一份非常全面的薄层TLC点板攻略
2022-03-25
-

QC, IQC, IPQC, QA,到底是什么鬼?
2020-05-27
-

AAS法分析茶叶中的铅,镉,砷
2020-05-27
-
红外光谱分析,你了解多少?
2021-01-11
-

超净工作台原理,使用与维护
2020-05-27
-

HPLC检测器,你了解吗?
2024-03-06
-

三聚氰胺,你还要害多少人
2020-05-27
-

一份写好的《方法确认作业指导书》, 直接参考使用
2021-04-25
-

选对色谱柱,快速开发方法
2020-05-27
-

'die','device','chip'有什么区别?
2024-02-21

加载更多