色谱分析中难搞的9种液相色谱峰
01
峰拖尾
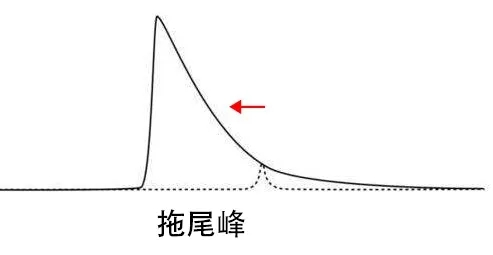
峰拖尾原因总结如下:
1、柱筛板堵塞:色谱柱的进出口的筛板堵塞,样品进入色谱柱时会受阻,液体流动受阻形成延迟,使得样品在相中停留时间变长,从而使峰型拖尾。
阻塞原因:A、配置流动相时污染 B、型号规格插口不匹配,在扭紧的那时候造成形变而促使管道阻塞。C、试品解决液清洁得不整洁,长期性会在六通阀和柱中间产生堵塞受阻。D、在应用手动式六通阀时,旋转的不及时,因此导致流路产生了死堵,工作压力迅速上升超出警示值。E、应用较高浓的缓存盐溶液,在关机时将会在出入口端结晶体成块并导致阻塞。
解决办法:需要通过反冲色谱柱,或者更换筛板。
2、色谱柱塌陷:是指色谱柱由于其它原因引起了柱效率丧失,不能对 物质形成保留,使得物质不在固定相上保留而随流动相流出,但是又还 有一点柱效,因此形成拖尾。
色谱柱坍塌分为两种:柱头塌陷和相塌陷。
(1)柱头塌陷:是指色谱柱使用较长的一段时间后,由于填料中硅胶基质或键合相的流失,导致柱床松动,在单向压力的作用下(色谱柱入口端承受绝大部分系统压力而出口端压力很小),整体表现为色谱柱的入口端出现填料缺失,严重时可看到填料的缺口。
柱头塌陷原因:
A.由流动相而不是由样品引起的,因为样品的进样量很小,对填料损伤有限,不至于造成那么大量的硅胶基质和键合相的流失,而流动相的量很大,1.0ml/min的流速,如果pH超标或在色谱柱pH耐受范围临界点附近长时间使用,是比较容易造成柱头塌陷的。
B.硅胶基质的溶解规律是:a、纯水相的流动相中硅胶的溶解度远大于带有一定比例有机相的流动相;b、流动相呈碱性比呈酸性更容易溶解硅胶基质,pH>9的时候硅胶溶解显著;c、温度越高硅胶越容易溶解,一般最好控制在40℃以下。
解决办法:通常无法修复。
(2)相塌陷:是指常规C18柱长时间用高含水量的流动相(>90%)长时间冲洗后,导致保留时间逐渐变短,最后可能在色谱柱上一点保留也没有的现象。
相塌陷原因:
在高含水量的流动相条件下,由于C18长链不溶于水,长时间冲洗时,键合上去的C18长链慢慢的相互聚集形成单独的一相,水也单独成一相,因此化合物在流动相的带动下越来越难与C18长链相互作用实现保留,最后一点保留都没有就直接在死体积的位置被冲洗出来。
解决办法:色谱柱出现了相塌陷后,通常用纯乙腈或甲醇持续冲洗可以使出现相塌陷的色谱柱恢复到原来的状态,但完全恢复需要比较长的时间,通常30分钟到1个小时是必须的。有一种更好的解决办法是用丙酮做流动相进行冲洗,所需时间更短、效果更好。
3、色谱柱污染:即样品不在同一起跑线起跑,从后面开始跑得到达终点稍晚,表现出拖尾。
污染原因:来源于流动相污染及仪器长时间未清洗导致色谱柱的污染。
解决办法:更换色谱柱或者采用有机溶剂梯度洗脱1h以上冲洗柱子。
4、流动相PH值选择错误:如某PH下有的样品存在分子型和离子型的动态平衡,或者两种形态比例接近1:1时,离子型的陆续向分子型转化就会表现出拖尾。
原因:不适宜的流动相PH。
解决办法:调节PH值可抑制分子解离,改善拖尾.对于碱性化合物,可以选用相对较低的PH值更有利于得到对称峰,也可以选用碱性试剂与固定相发生反应,从而阻断碱性化合物与固定性发生反应。
02
峰前沿

前沿峰原因总结如下:
1、样品过载:被保留的样品在正常出峰时间前陆续出来,形成前沿峰。降低样品含量。
原因:
(1)色谱柱载量不够;
(2)样品浓度过大;
(3)进样量太大
解决办法:从上述三方面进行优化。
2、样品溶剂选择不恰当:在反相色谱中用已腈做样品溶剂,而流动相的洗脱力较弱时会出现前沿峰。选择流动相或者接近流动相的比例作为样品溶剂。
原因:溶剂的洗脱能力大于流动相
解决办法:(1)选择合适的溶剂溶解样品;(2)降低进样量;(3)调整流动相的有机比例。
3、色谱柱损坏:色谱柱损害严重,不能对化合物形成保留。
原因:色谱柱使用后未及时清洗等
解决办法:更换色谱柱。
4、包峰:由于分析方法不适宜导致在主峰前有很多小峰出现,假象前沿峰(非其他因素引起的前沿),大峰包埋了没有分开的小峰。
解决办法:优化分析方法。
03
形成倒峰
倒峰原因总结如下:
1、样品折射率小:示差折光检测器测的信号是折射率小的物质。
原因:
(1)组分的折射率小于流动相。(通常是水引起的)
(2)密度不一样,出来的峰正倒不一样。
解决办法:通常不需要解决。
2、进样所致:进样时,样品对原有流动相产生瞬间阻断,然后瞬间涌流,所以产生先倒后高的“溶剂峰”。
原因:断流
解决办法:不需要解决。
04
包裹峰/严重拖尾峰
包裹峰/严重拖尾峰原因总结如下:
1、色谱柱问题:色谱柱基质流失,或者色谱柱被真菌污染。
解决办法:更换色谱柱。
2、样品未全部溶解或者溶解度低:温度、溶解都可能会影响到溶解度。
原因:室温过低、样品溶解性差。
解决办法:选择合适溶解来溶解样品。
3、流动相混合不均匀:一般是流动相没配好,管路流动相不均匀,也会导致上述现象。
解决办法:
(1)充分混合流动相,如果组分较复杂,手动混匀时间、超声脱气时间可适当延长。(2)两项或多项在仪器内无法混匀时,可选择手动提前混匀,该流动相不可以选用仪器进行混合。
05
基线向上或向下漂移
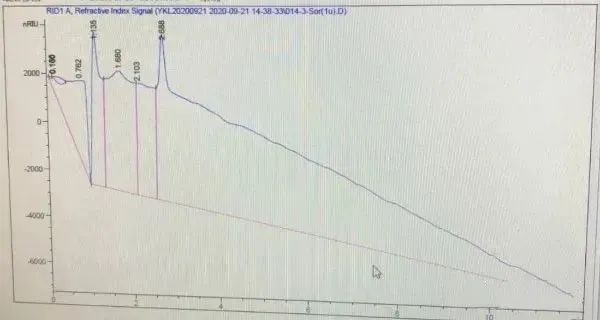
基线向上或向下漂移原因总结如下:
1、色谱柱柱温不稳定:色谱柱柱温未稳定会导致示差检测器、电导检测器、较低灵敏度的紫外检测器或其它光电类检测器产生波动,因此会导致基线的飘逸。
原因:柱温波动
解决办法:
(1)选择可以控温的柱温箱;
(2)增加柱温平衡时间,一般为30min;
(3)在检测器之前使用热交换器。
2、流动相中有机相和水相为完全混匀:流动相条件变化引起的基线漂移大于温度导致的漂移。
解决办法:
(1)充分混合流动相,如果组分较复杂,手动混匀时间、超声脱气时间可适当延长。(2)两项或多项在仪器内无法混匀时,可选择手动提前混匀,该流动相不可以选用仪器进行混合。
(3)使用HPLC级的溶剂,流动相在使用前进行脱气处理。
3、流通池被污染或有气体:用甲醇或其他强极性溶剂冲洗流通池。
解决办法:
(1)排气。
(2)用甲醇、乙腈或其他极性强的有机溶剂,也可以用0.1N的硝酸(不要用盐酸),进行清洗流通池。
4、流动相配比不当或流速变化:更改流动相配比或流速。
解决方法:可定期检查流动相组成及流速。
5、样品中有强保留的物质:以馒头峰样被洗脱出,从而表现出一个逐 步升高的基线。
解决办法:1使用保护柱,2如有必要,在进样之间或在分析过程中, 定期用强溶剂冲洗柱子。
6、波长的设置问题:低波长时,由于有机溶剂的比例增加,随之基线也会发生漂移, 高波长时, 基线相对平稳。
解决办法:设置高波长。
06
峰展宽
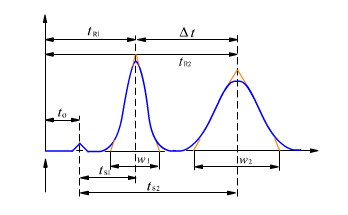
峰展宽原因总结如下:
1、色谱柱污染或柱校降低 色谱柱的污染会造成塔板数降低,引起峰展宽现象。
解决办法:更换同样类型的色谱柱,如果新柱子可以提供对称的色谱峰,则用强溶剂冲洗旧柱子。
2、柱子与检测器之间的管路太长或管路内径太大会引起宽峰现象
解决办法:更换内径较小的短管路。
3、检测器或流通池体积过大:
解决办法:减少响应时间或使用更小的流通池。
4、溶剂洗脱能力比较低
原因:流动相洗脱力不足导致化合物保留时间延长引起宽峰。
解决办法:选用洗脱力强的溶剂:乙腈、四氢呋喃等。
07
噪音过大
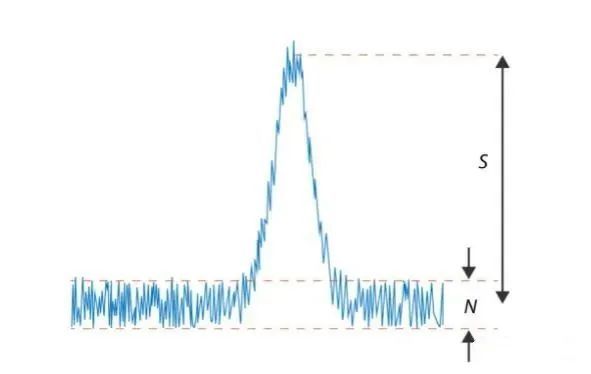
噪音过大原因总结如下:
1、在流动相、检测器或泵中有气泡
解决办法:流动相脱气,冲洗系统 以除去检测器或泵中的空气。
2、系统漏液:检查管路接头是否松动,泵是否漏液,是否有盐析出和不正 常的噪音。
解决办法:检查色谱柱是否有漏液,检查单向阀是否有盐洗出,检查流动相瓶中过滤头是否有持续的小气泡,检查检测器流出管路是否有气泡,如有必要,更换泵密封。
3、流动相没混匀
解决办法:用手摇动使混合均匀或使用低粘度的溶剂,增加超声时间。
4、柱温异常、检测器温度异常
解决办法:使用柱温箱,减少温度差异 或加上热交换器。
5、偶然噪声: 进行断电检查,液相、检测器等,追溯干扰的外部来源,加以调整。
解决办法:采用精密级稳压电源。
08
色谱峰之间分离度降低
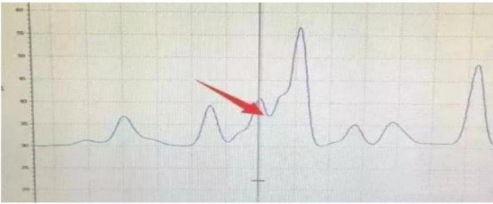
色谱峰之间分离度降低原因总结如下:
1、分析方法梯度洗脱程序不合理:快速洗脱往往会使分离度不够。
解决办法:优化梯度洗脱程序,采用缓慢洗脱方式,要么延长时间,要么降低流速。
2、流动相微生物污染:
解决办法:含有有机铵盐-乙酸的流动,通常需要现用先配,长时间不用会导致PH或者盐浓度发生变化,降低分离度,需重新配置流动相。
3、保护柱或分析柱阻塞:
解决办法:
(1)更换新的保护柱进行分析,
(2)如果是分析柱阻塞,更换色谱柱。
09
分叉峰
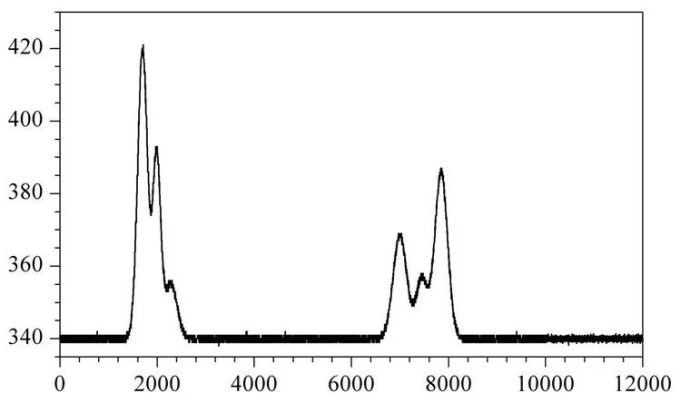
分叉峰原因总结如下:
原因:
1、样品或色谱柱污染。
2、溶剂效应
3、色谱柱过载。
3、色谱柱塌陷
4、色谱柱两个接头没有安装好。
5、色谱柱性能下降。
6、柱前筛板部分堵塞
解决办法:从上述原因,逐个排除。
药品研发驿站
展源
何发
相关文章
-

选对色谱柱,快速开发方法
2020-05-27
-

AAS法分析茶叶中的铅,镉,砷
2020-05-27
-

色谱峰裂分,前拖尾的诊断
2020-05-27
-

检测有机氯类农药,气相色谱法检测法
2021-01-12
-
优化色谱分析流通量
2020-05-27
-

色谱分析流通量优化
2020-05-27
-

色谱分析介绍及色谱制备的方法
2021-05-12
-

最全色谱分析鬼峰
2024-05-30
-

色谱分析大起底,色谱概论经典知识!
2022-02-23
-

白酒香味气相色谱分析
2020-09-27

加载更多