【攻略】最详尽的TLC技术深度解析!
TLC技术
薄层色谱法(TLC),系将适宜的固定相涂布于玻璃板、塑料或铝基片上,成一均匀薄层。待点样、展开后,根据比移值(Rf)与适宜的对照物按同法所得的色谱图的比移值(Rf)作对比,用以进行药品的鉴别、杂质检查或含量测定的方法。薄层色谱法是快速分离和定性分析少量物质的一种很重要的实验技术,也用于跟踪反应进程。关于TLC的那些技巧快来看看吧~
原理:
利用吸附剂(硅胶)对样品中各组分吸附能力不同,即展开剂对它们的解吸附能力的不同,是各组分达到分离的目的。
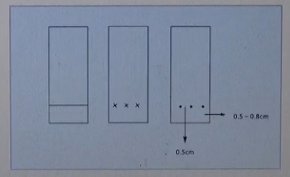
遵守原则:点样的高度一定要高于展开缸中展开剂的高度原点间距0.3-0.5cm,底边距离0.5-0.8cm即可
(1)一般机型溶剂体系(CH2Cl2,THF,ethyl acetate,etc
)可以直接取样
,也可以稀释后取样(如果反应体系浓度太大,需要适当稀释后再TLC检测)
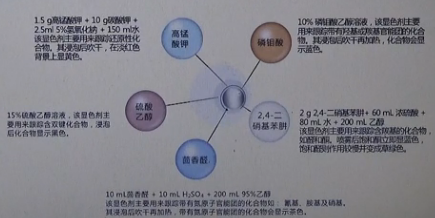
(2)强极性溶剂体系(如DMSO, DMF),不适宜直接取样进行TLC检测
,最好简单后处理(加水和有机溶剂萃取)以后取样。因为DMF,DMSO的沸点很高,不易吹干,再加上它们的极性比较大,如果直接取样进行TLC检测的话,就会导致整个展开剂的极性增大,会使展开效果大打折扣,特别是对于一些本身极性比较接近的化合物,更不利于极性大小的判断,所以碰到这种反应类型,通常处理办法是取一些样品放于离心管内,再往离心管中加水和乙酸乙酯,震荡几下后放置,取乙酸乙酯相进行TLC检测,这样通过简单的萃取过程可以把样品萃取到有机相中,而DMF或DMSO会被留在水相内
(3)需要猝灭的体系(NaH, LiAlH4,n-BuLi, etc),需处理(如加水或酸和有机溶剂)后取样NaH, LiAlH4, n-BuLi, etc反应完后会以盐的形式存在,没办法直接取样进行检测,需要简单后处理后,再进行检测。具体的操作是取些样品放于离心管内,加水或酸淬灭后,再加入有机溶剂,震荡后再进行TLC检测
(4)强碱、酸性物质(NaOH, H2SO4, etc),最好中和后取样
Eg. 含吡啶结构的化合物,在强酸性体系内会成盐,显然直接取样进行TLC检测是不合适的,这是就需要中和后,在进行检测,判断。
需要注意:1.产品在水相还是有机相;2.有没溶解固体
(1)后处理有机层产物很少看看是否产物极性太大,水溶性太好
,有机层水层同时点板取样进行TLC检测时,确实对反应进行简单的后处理,但发现萃取后有机相没有要的东西,或东西很少,碰到这种情况,回头再检测水相,看东西是否在水相中,没被萃取到有机相中,因为碰到一些极性较大的化合物时,它们在水相中的溶解度很好,就不会很容易萃取带有机相了,所以我们在TLC检测时,最好是有机相和水相点在同一块板上,同时检测来综合比较。
把没有溶解的固体取出后找合适的溶剂溶解后再跟溶解的部分共同TLC检测比较来综合判断反应进行的情况在用TLC 跟踪反应时,发现反应体系里有不溶解的固体,这种情况,很多时候可能会把它忽略掉,直接只取溶解的反应液直接跟踪,这种操作方法是不正确的,有时候固体就是产物,如果只取反应液检测,就会得出与实际情况不符的结论,导致我们判断失误。还有一些反应是原料在反应体系中溶解度不好,但是产物溶解度比较好,TLC跟踪时如果只点反应液的话,可能会认为原料已经反应完了,就草草的把反应处理掉,但实际上原料还剩下很多,所以建议大家碰到这种情况时,取出一些固体,找其他合适的溶剂溶解后,再和反应液一起来进行TLC检测,综合判断反应进行的情况
(3)Boc等保护氨基脱Boc保护成盐情况,如何分析
这种反应一般是在盐酸甲醇,盐酸乙酸乙酯等酸性体系中进行,那么反应完成后,生成的胺在这种酸性体系中无法以游离的形式存在,而是以盐酸盐的形式存在,所以在TLC跟踪反应时,如果发现原料已经反应完全,而在原点有一个很大的斑点的话,通常是可以得出反应已经进行完全的结论了
酰氯的活性很高,直接取样进行TLC检测的话,可能会碰到空气中的水汽又会使其变回羧酸所以直接进行TLC检测并不可行,我们可以利用它活性高的特点,可以去一些反应液出来溶解到甲醇或甲胺类物质中,这样反应液就会与甲醇或甲胺类物质反应酯或酰胺,然后借用TLC或LCMS等检测手段,通过检测生成的酯或酰胺来判断反应进行情况
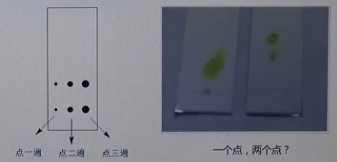
a. 点样的浓度要控制适当。如果点样太浓的话,特别是对于极性相差不大的化合物,就会导致它们的分离度变差,出现两个点重合成一个点的现象,同样点样浓度太稀,也不利于检测判断。
c. 0.3mm毛细点有机相,0.5mm点水相较方便。
a.对所需组分有良好的溶解性;
b.可使各组分间有较好的分离;
c.待测组分的Rf 在0.2~0.8之间;
d.不与待测组分发生化学反应;
e.沸点适中,黏度较小;
石油醚<二氯甲烷<乙醚<四氢呋喃<乙酸乙酯<丙酮<正丙醇<甲醇<水
1. 一般极性的化合物:PE(石油醚)/EA(乙酸乙酯)体系
2. 极性较大的化合物:DCM(二氯甲烷)/CH3OH体系
可以通过调节两种溶剂的配比来调节展开剂的极性大小,对于一般极性的化合物,通常,用石油醚乙酸乙酯体系就可以,如果化合物的极性很大,用纯乙酸乙酯Rf值都很小时,就要选择二氯甲烷甲醇体系了。
展开剂体系的选择原则:
脂肪类胺化合物:在用硅胶板展开时,由于硅胶的弱酸性作用,比较容易出现拖尾的状况,那么以在展开剂中加入0.1%(约为1滴)的氨水或三乙胺混合均匀后再展开,能比较的减少或抑制拖尾情况
在展开剂中加入微量的醋酸(乙酸或甲酸的作用用,一方面增大展开剂极性, 另外也可以抑制硅胶上羟基的作用,减少拖尾)另外常用的混合体系:
PE/DCM, PE/Acetone(丙酮),EA/DCM, EA/CH3OH
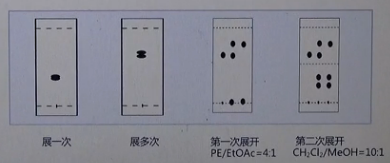
展开方向有单向、单向长版、多次、多种极性展开等。
最常用的是单次展开,如果展开一次后两个斑点离的很近,可以选择换一块长板,或多次展开,如果反应体系中的物质极性跨度比较大,可以选择多种极性展开的方式,即可以将TLC板放入石油醚乙酸乙酯体系中,先将极性较小的化合物展开,拿出来吹干后,再放入大极性的二氯甲烷甲醇体系中,将大极性的物质展开。
一般以目标产物的Rf值在0.4-0.6左右为最佳;
一般选用展开剂极性应从低到高原则。
展开后的点位置偏高或偏低较多,需要重新调整展开剂的配比,或者换展开剂体系
一看:
首先在日光下观察,划出有色物质的斑点位置。
如一些含硝基的化合物,在日光下就可以看到黄色的斑点
三碘:
对于紫外吸收很弱的物质,可以用碘缸进行显色,利用大部分有机物会吸附碘可逆的产生棕色或黄色斑点,用碘缸进行显色时,需要注意以下两点:TLC板在放入碘缸前,需将溶剂挥发干或者吹干;某些胺类化合物如三乙胺、DLEA等在碘缸中也会显色,所以,如果反应体系中用到这些试剂的话,要注意判断
四显:
既无色又无紫外吸收且在碘缸中不显色的物质,可以用显色剂显色
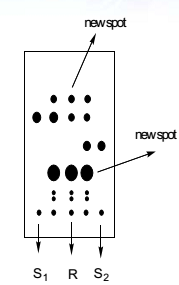
反应有无新点产生;原料有无剩余,剩余多少;反应是否干净;综合判断反应如何处理。
2. 监测时间,对于需要过夜的反应,建议在过夜前检测反应跟过夜后的反应检测作对比,从而决定下一次反应是否有过夜的必要,那么对于某些速度很快的反应,可以将检测反应的间隔时间尽量缩短一些;
3. 留样作对照,如果小试已经拿到标准点的话,我们可以留一些样品,作为放大反应的标样使用;
4.当碰到一些已经摸索了很多条件,反应效果都不太好的情况下时,建议将重要板留存,把不同反应条件下的TLC板综合比较,可能会得到一些有用的信息。跟踪监测反应时还有一点需注意,在TLC检测时,除了点原料点和反应液点外,最好同时点原料和反应液的交叉点,这样板展开后更利于分析判断
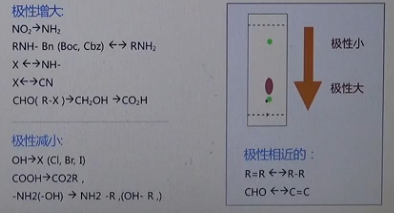
对于像硝基还原为氨基,或醛还原为醇的反应,我们看到这些反应产物的极性比原料的极性大,那么在用硅胶板跟踪反应时,如果发现原料已经消耗完,而在原料点的下方有主要新点生成的话,大致可以得出反应应该是朝预期的方向进行的结论;
相反,对于极性减小的反应,如胺类化合物上保护基等反应,如果TLC检测发现在原料点的上方有主要的新点生成的话,也能得到类似的结论,但是还有一些反应,如烯烃的还原,这种反应的原料和产物的极性很接近,可能单纯的凭借TLC板,不是很好判断反应的进行情况,那么这是就需要借助其他的检测手段来综合判断了
TLC- - HPLC/LC- - Ms联用,可以用于对反应进行定性和定量分析,确定反应有目标产物后,可以将TLC-- MS联用,确定标样点
。
具体操作:
在一块TLC小板上,上一条样品带,展开后将不同的色带分别刮取后,用溶剂溶解,注射器加滤头过滤掉硅胶后,将溶液用MS检测,确定标样点,如果出现两条色带,MS值相同,有异构体的情况,还可以将TLC-- NMR联用来确认结构
在跟踪萃取的过程中,不能单纯根据有机相有没有颜色或水相有没有紫外吸收来判断,因为可能萃取的次数越多,反而是被萃取到有机相的杂质越多,所以建议在萃取2-3次后可以点一块TLC板,并一定要把TLC板展开后再来判断是否继续萃取。同理,重结晶过程也是如此
固体的纯度和母液的取舍也是要把TLC板展开后再下结论
a. Rf值在0.2左右的展开剂比例为过柱子时的洗脱剂的合适值
b. 由于溶解度的原因,有时过柱时组分流出顺序未必和TLC上点极性顺序一致
有时做了小试后,想拿着标样点或者量少的产品进一步纯化时,不一定非要过柱子,也可以选择制备TLC来进行,通常比较快速,而且分离效果比较好。
6、制备TLC顺序:点板--展开--显色--刮板--洗脱
a. 溶剂:极性小,溶解性好;尽量选择极性小,挥发性好,对样品溶解性好的溶剂,如二氯甲烷;
b. 上样量:40-60mg;如果反应不太干净,可适当再减少;c. 上样位置:距离两端大约1cm,距离底边大约2cm,原则是点样的高度一定要高于展开缸中展开剂的高度
b. 所用展开剂的极性较展小板时大,如展小板时展开剂的比例石油醚乙酸乙酯是4:1的话,再用制备TLC时可将展开剂的比例扩大到石油醚乙酸乙酯2:1
上样时如果上的色带太宽或不齐,通常直接展开的话,展开效果不是太好,不利于去分析判断,那么我们可以在枪头中塞一点棉花,前端用剪刀剪平整后再去上样,通常色带就会比较平整
如果出现上述情况,还可用大极性溶剂预展开2-3cm,来尽量抑制产生的不良影响,具体来讲,如果要用石油醚乙酸乙酯为4:1的展开剂来展开,我们可以先将TLC板放入石油醚乙酸乙酯为1:1的展开剂中,展开2-3cm后取出来,色带就会相对比较平整,将溶剂吹干后再放入石油醚乙酸乙酯为4:1的展开剂继续展开
需要用碘或显色剂显色的时候,我们将制备的一侧进行部分显色,活用玻璃刀将TLC板分割下小部分,通过部分的显色情况,来反推整块板的显色情况,所以相对的对板的展开效果的要求比较高
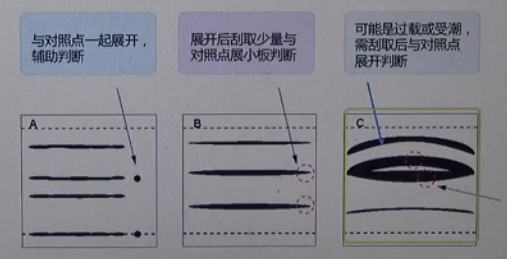
如果有标样点的话,可以跟标样点TLC检测对照;如果没有标样点的话可以将TLC—LCMS或NMR组合判断;有时在大板展开后,可能会出现像第3块板的情况,两条色带距离特别近,不好判别是不是一个东西,造成这种状况的原因可能是过载或受潮,建议大家将两条色带分别刮取后与标点对照来展开判断是不是一个东西,再确定能不能合并
刮板:
是将不同的色带刮取后放入不同锥形瓶中进行洗脱
Note:刮板后,在没有得到确定的结论前不能轻易丢掉薄板
洗脱:
尽量选择溶解度好,极性小的溶剂,通常可用二氯甲烷或者乙酸乙酯,如果化合物的极性很大用纯的二氯甲烷或乙酸乙酯洗脱效果不好时,也可选用二氯甲烷比甲醇等于8:1或乙酸乙酯比甲醇等于10:1的混合溶剂进行洗脱,洗脱剂应避免加入过多的甲醇,以免产物中混有大量硅胶,另外,如果需要MS检测,溶解的硅胶析出后会造成柱子的堵塞。

制药人资源
展源
何发
















加载更多