作者曾经接触过不少刚从事GC工作的技术人员,他们能很快掌握必要的GC基础知识,甚至能很快操作仪器。但当接到一个分析任务时,面对样品不敢确定从何做起。也有一些人认为GC分析很简单,不就是打一针就可得到结果吗。其实不然!就如医院的护士打针,你若不了解病人情况,不知道用药的剂量,随便给病人打一针就能治病吗?
这就涉及到了方法开发问题。简单地说,方法开发就是针对一个或一批样品建立一套完整的分析方法。就GC而言,就是首先确定样品预处理方法,然后优化分离条件,直至达到满意的分离结果。最后建立数据处理方法,包括定性鉴定和定量测定。当然,这一方法要真正成为实用方法,还必须进行验证。下面首先讨论方法开发的一般步骤。
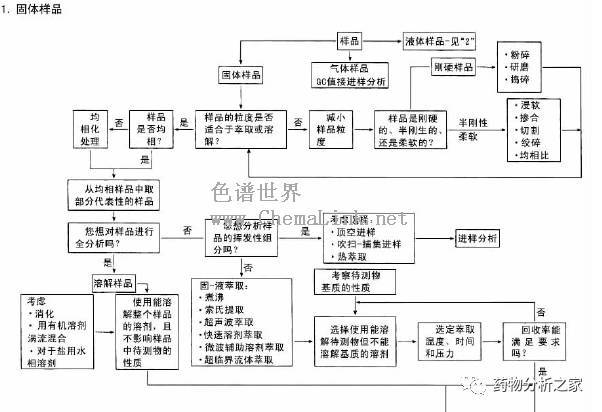
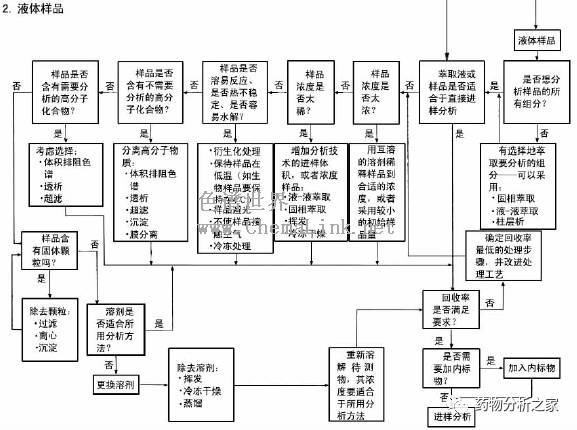
GC能直接分析的样品必须是气体或液体,固体样品在分析前应当溶解在适当的溶剂中,而且还要保证样品中不含GC不能分析的组分(如无机盐)或可能会损坏色谱柱的组分。这样,我们在接到一个未知样品时,就必须了解它的来源,从而估计样品可能含有的组分,以及样品的沸点范围。如能确认样品可直接分析,问题就简单了。只要找一种合适的溶剂,如丙酮、己烷、氯仿、苯等就是GC常用的溶剂。一般讲,溶剂应具有较低的沸点,从而使其容易与样品分离。尽可能避免用水、二氯甲烷和甲醇作溶剂,因为它们对延长色谱柱的使用寿命不利。另外,如果用毛细管柱分析,应注意样品的浓度不要太高,以免造成柱超载,通常样品的浓度为mg/ml级或更低。
如果样品中有不能用GC直接分析的组分,或者样品浓度太低,就必须进行必要的预处理,包括采用一些预分离手段,如各种萃取技术、浓缩方法、提纯方法等。下面给出一个简要的样品预处理指南(见表5-1-4),表5-1-4 样品处理指南有关各种处理方法的详细信息,请读者参看《色谱分析样品处理》分册。
需要强调的一点是,无论样品处理方法,还是下面要讨论的GC分析条件确定,文献调研都是很重要的方法开发步骤。所以,开始实验前,应当查一下文献。若文献中已有相同样品的分析方法,那就会大大加快方法开发的过程。只要在此基础上作一些必要的优化即可。即使能找到类似样品的分析方法,也可以作为重要的参考,从而避免走一些不必要的弯路。
所谓仪器配置就是用于分析样品的方法采用什么进样装置、什么载气、什么色谱柱以及什么检测器。比如,要用GC分析啤酒的挥发性成分,就需要一个顶空进样器;要测定水中痕量含氯农药的残留量,就要用电子俘获检测器。就色谱柱而言,常用的固定相有非极性的OV-1(SE-30)、弱极性的SE-54、极性的OV-17和PEG-20M等。可根据极性相似相容原理来选用,即分离一般脂肪烃类(如柴油或汽油)时多用OV-1(SE-30),分析醇类和酯类(如含酒精饮料)多用PEG-20M,分析农药残留量则多用OV-17或OV-1701。而要分析特殊的样品,如手性异构体,就需要特殊的色谱柱。对于很复杂的混合物,SE-54往往是首选的固定相。
当样品准备好,且仪器配置确定之后,就可开始进行尝试性分离。这时要确定初始分离条件,主要包括进样量、进样口温度、检测器温度、色谱柱温度和载气流速。
进样量要根据样品浓度、色谱柱容量和检测器灵敏度来确定。样品浓度不超过12 3 14 时填充柱的进样量通常为1-5μL,而对于毛细管柱,若分流比为50:1时,进样量一般不超过2μL。如果这样的进样量不能满足检测灵敏度的要求,可考虑加大进样量,但以不超载为限。必要时先对样品进行预浓缩,还可考虑采用专门的进样技术,如大体积进样,还可采用灵敏度更高的检测器。
进样口温度主要由样品的沸点范围决定,还要考虑色谱柱的使用温度。即首先要保证待测样品全部汽化,其次要保证汽化的样品组分能够全部流出色谱柱,而不会在柱中冷凝。原则上讲,进样口温度高一些有利,一般要接近样品中沸点最高的组分的沸点,但要低于易分解组分的分解温度,常用的条件是250-350℃。大多数先进GC仪器的进样口温度均可达到450℃。这时,沸点为+**7左右的组分均可汽化(因为在溶液状态下,组分的沸点会降低一些)。实际操作中,进样口温度可在一定范围内设定,只要保证样品完全汽化即可,而不必进行很精确的优化。注意,当样品中某些组分会在高温下分解时,就用适当降低汽化温度。必要时可采用冷柱上进样或程序升温汽化(PTV)进样技术。
色谱柱温度的确定主要由样品的复杂程度和汽化温度决定。原则是既要保证待测物的完全分离,又要保证所有组分能流出色谱柱,且分析时间越短越好。组成简单的样品最好用恒温分析,这样分析周期会短一些。特别是用填充柱时,恒温分析时色谱图的基线要比程序升温时稳定得多。对于组成复杂的样品,常需要用程序升温分离,因为在恒温条件下,如果柱温较低,则低沸点组分分离得好,而高沸点组分的流出时间会太长,造成峰展宽,甚至滞留在色谱柱中造成柱污染;反之,当柱温太高时,低沸点组分又难以分离。图5-1-3给出了一个示意图。说明程序升温的必要性。实际上,毛细管柱的一个最大优点就是可在较宽的温度范围内操作这样既保证了待测组分的良好分离,又能实现尽可能短的分析时间。
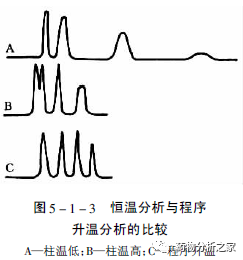
一般来讲,色谱柱的初始温度应接近样品中最轻组分的沸点,而最终温度则取决于最重组分的沸点。升温速率则要依样品的复杂程度而定。在没有资料可供参考的情况下,建议毛细管柱的尝试温度条件设置为:
 注意,这只是方法开发时的初始参考条件,具体工作中一定要根据样品的实际分离情况来优化设定。
检测器的温度是指检测器加热块温度,而不是实际检测点,如火焰的温度。检测器温度的设置原则是保证流出色谱柱的组分不会冷凝,同时满足检测器灵敏度的要求。大部分检测器的灵敏度受温度影响不大,故检测器温度可参照色谱柱的最高温度设定,而不必精确优化。比如在使用OV-101或OV-1毛细管色谱柱时,火焰离子化检测器(FID)的温度可设定为300℃。
载气流速的确定相对容易一些,开始可按照比最佳流速(氮气约为20cm/s,氦气约为25cm/s,氢气约为30cm/s)高10%来设定。然后再根据分离情况进行调节。原则是既保证待测物的完全分离,又要保证尽可能短的分析时间。用填充柱时,载气流速一般设为30ml/min。
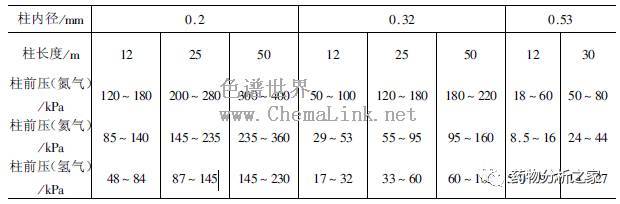
需要指出的是,当仪器没有配置电子气路控制(EPC)时,必须通过皂膜流量计或测定死时间的方法来测定载气流速,通过调节往前压的方式来改变载气流速。色谱柱越长,内径越小,柱温越高,需要柱前压越高。表5-1-5给出了常用毛细管色谱柱采用的柱前压参考数值范围。初始条件一般设置为表中给定范围的上限,然后根据实际分离情况再进行优化。
此外,当所用检测器需要燃烧气和/或辅助气时,还要设定这些气体的流量。有关检测器的说明书通常会列出适合的气体流量值,可供参考。比如,用毛细管柱和FID时,检测器气体流量可设定为:
上述初始条件设定后,便可进行样品的尝试性分析。一般先分离标准样品,然后分析实际样品。在此过程中,还要根据分离情况不断进行条件优化。
注意,这只是方法开发时的初始参考条件,具体工作中一定要根据样品的实际分离情况来优化设定。
检测器的温度是指检测器加热块温度,而不是实际检测点,如火焰的温度。检测器温度的设置原则是保证流出色谱柱的组分不会冷凝,同时满足检测器灵敏度的要求。大部分检测器的灵敏度受温度影响不大,故检测器温度可参照色谱柱的最高温度设定,而不必精确优化。比如在使用OV-101或OV-1毛细管色谱柱时,火焰离子化检测器(FID)的温度可设定为300℃。
载气流速的确定相对容易一些,开始可按照比最佳流速(氮气约为20cm/s,氦气约为25cm/s,氢气约为30cm/s)高10%来设定。然后再根据分离情况进行调节。原则是既保证待测物的完全分离,又要保证尽可能短的分析时间。用填充柱时,载气流速一般设为30ml/min。
需要指出的是,当仪器没有配置电子气路控制(EPC)时,必须通过皂膜流量计或测定死时间的方法来测定载气流速,通过调节往前压的方式来改变载气流速。色谱柱越长,内径越小,柱温越高,需要柱前压越高。表5-1-5给出了常用毛细管色谱柱采用的柱前压参考数值范围。初始条件一般设置为表中给定范围的上限,然后根据实际分离情况再进行优化。
此外,当所用检测器需要燃烧气和/或辅助气时,还要设定这些气体的流量。有关检测器的说明书通常会列出适合的气体流量值,可供参考。比如,用毛细管柱和FID时,检测器气体流量可设定为:
上述初始条件设定后,便可进行样品的尝试性分析。一般先分离标准样品,然后分析实际样品。在此过程中,还要根据分离情况不断进行条件优化。
分离优化是一个很大的题目,有专门的优化理论来研究,市场上还有计算机软件可用于优化。本书不准备就此展开详细讨论,只是在下一节从实用的角度简单介绍优化方法。在这里只强调操作条件柱温和载气流速的优化。
事实上,当样品和仪器配置确定之后,一个色谱技术人员最经常的工作除了更换色谱柱外,就是改变色谱柱温和载气流速,以期达到最优化的分离。柱温对分离结果的影响要比载气的影响大。
简单地说,分离条件的优化目的就是要在最短的分析时间内达到符合要求的分离结果。所以,当在初始条件下样品中难分离物质对的分离度R大于1.5时,可采用增大载气流速、提高柱温或升温速率的措施来缩短分析时间,反之亦然。比较难的问题是确定色谱图上的峰是否单一组分的峰。这可用标准样品对照,也可用GC/MS测定峰纯度。如果某一感兴趣的峰是两个以上组分的共流出峰,优化分离的任务就比较艰巨了。在改变柱温和载气流速也达不到基线分离的目的时,就应更换更长的色谱柱,甚至更换不同固定相的色谱柱,因为在GC中,色谱柱是分离成败的关键。
所谓定性鉴定就是确定色谱峰的归属。对于简单的样品,可通过标准物质对照来定性。就是在相同的色谱条件下,分别注射标准样品和实际样品,根据保留值即可确定色谱图上哪个峰是要分析的组分。定性时必须注意,在同一色谱柱上,不同化合物可能有相同的保留值,所以,对未知样品的定性仅仅用一个保留数据是不够的。双柱或多柱保留指数定性是GC中较为可靠的方法,因为不同的化合物在不同色谱柱上具有相同保留值的几率要小得多。对于复杂的样品,则要通过保留指数和/或GC/MS来定性。事实上,GC/MS是当今GC定性的首选方法,它可以给出相应色谱峰的分子结构信息,同时还能做定量分析。不过,我们应当了解,GC/MS并不总是可靠的,尤其是一些同分异构体,它们的质谱图往往非常相似,故计算机检索结果有时是不正确的。只有当GC保留指数和MS图的鉴定结果相吻合时,定性的可靠性才是有保障的。
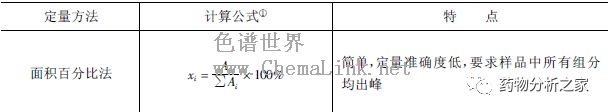
在这一步骤是要确定用什么定量方法来测定待测组分的含量。常用的色谱定量方法不外乎峰面积(峰高)百分比法、归一化法、内标法、外标法和标准加入法(又叫叠加法)。表5-1-6列出了这些方法的原理和特点。
峰面积(峰高)百分比法最简单,但最不准确。只有样品由同系物组成、或者只是为了粗略地定量时,该法才是可选择的。当然,在有机合成过程中监测反应原料和+ 或产物的变化时,也可用此法作相对定量。
因为不同的化合物在同一条件下、同一检测器上的响应因子(单位峰面积代表的样品量)往往不同,故须用标准样品测定响应因子进行校正后,方可得到准确的定量结果。其他几种定量方法均需要校正。相比起来,归一化法较为复杂,它要求样品中所有的组分均出峰,且要求有所有组分的标准品才能定量,故很少采用。外标法是采用最频繁的方法,只要用一系列浓度的标准样品作出工作曲线(样品量或浓度对峰面积或峰高作图),就可在完全一致的条件下对未知样品进行定量分析。只要待测组分出峰且分离完全即可,而不考虑其他组分是否出峰和是否分离完全。需要强调,外标法定量时,分析条件必须严格重现,特别是进样量。如果测定未知物和测定工作曲线时的条件有所不同,就会导致较大的定量误差。还应注意,外标工作曲线最好与未知样品同时测定,或者定期重新测定工作曲线,以保证定量准确度。
相比而言,内标法的定量精度最高,因为它是用相对于标准物(叫内标物)的响应值来定量的,而内标物要分别加到标准样品和未知样品中。这样就可抵消由于操作条件(包括进样量)的波动带来的误差。与外标法类似,内标法只要求待测组分出峰且分离完全即认可,其余组分则可用快速升高柱温使其流出或用反吹法将其放空,这样就可达到缩短分析时间的目的。尽管如此,要找一个合适的内标物并不总是一件容易的事情,因为理想的内标物的保留时间和响应因子应该与待测物尽可能接近,且要完全分离。此外,用内标法定量时,样品制备过程要多一个定量加入内标物的步骤,标准样品和未知样品均要加入一定量的内标物。因此,只要定量精度要求不高,就应避免使用内标法。
至于标准加入法,是在未知样品中定量加入待测物的标准品,然后根据峰面积(或峰高)的增加量来进行定量计算。其样品制备过程与内标法类似,但计算原理则完全是来自外标法。标准加入法的定量精度应该介于内标法和外标法之间。
上述各种定量方法中,峰面积均可用峰高代替。理论上讲,浓度型检测器(如TCD,参见《气相色谱检测方法》)用峰高定量较准确,而质量型检测器用峰面积定量更准确。但在实际操作中,影响峰高和峰面积的因素有多种,不仅载气流速和柱温,而且检测器的结构设计等均有影响。综合考虑各种因素,峰面积定量一般比峰高定量准确一些。然而,当峰未完全分离时,面积积分准确度会下降,此时用峰高定量不失为一种合理的选择。
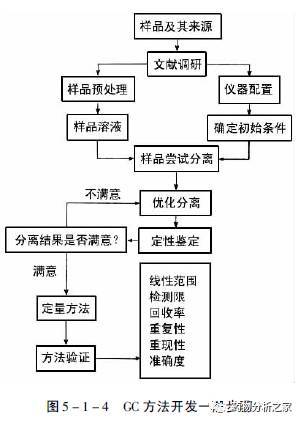
到此为止,我们基本上完成了一个GC方法的开发。这个方法是否合理、可靠?是否能为同行所采用?还有待于对其进行验证。图5-1-4总结了GC方法开发的一般步骤。



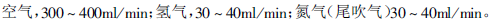















加载更多