常用的晶型检测方法你都知道哪些呢?本文整理了相关知识点为大家答疑解惑,跟小析姐一起来看看吧~
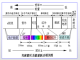
分析技术 |
测试指标 |
X射线衍射 |
粉末衍射XRPD、单晶RRD |
晶型,结晶度 |
热技术 |
差示扫描量热仪DSC、热重分析TGA、热显微镜(HSM) |
熔点,晶型,脱水脱溶剂温度 |
光谱 |
光学显微镜 |
粒径,晶癖 |
拉曼光谱Raman |
晶型,溶剂成分,溶液浓度,溶剂残留量 |
近红外NIR |
溶剂残留量,水分含量,转晶程度 |
固态核磁共振光谱 |
溶剂残留量,晶型 |
扫描隧道显微镜法 |
晶型(晶体表面单个原子及其排列状态) |
其他 |
动态水吸附DVS |
吸湿性 |
现对X射线衍射法、差示扫描量热法(DSC)、热重分析(TGA)检测原理及检测过程中可能遇到的问题进行简单描述。
一、Ⅹ射线行射法:
光在传播过程中,遇到障碍物或小孔时,光将偏离直线传播的途径而绕到障碍物后面传播的现象,叫光的衍射(diffraction of light)。
1、原理
光是一种电磁波,而X射线也是一种电磁波,其波长很小(约在0.001~100 nm)。当一束单色X射线入射到晶体时,由于晶体是由原子规则排列成的晶胞组成,这些规则排列的原子间距离与入射X射线波长有X射线衍射分析相同数量级,故由不同原子散射的X射线相互干涉,在某些特殊方向上产生强X射线衍射,衍射线在空间分布的方位和强度,与晶体结构密切相关,每种晶体所产生的衍射都反映出该晶体内部的原子分布规律。
X射线衍射的满足条件:布拉格方程 2dsinθ=nλ,其中,d为晶面间距,θ为入射线,反射线与反射晶面之间的夹角,λ为波长,n为反射级数,布拉格方程是X射线在晶体产生衍射时的必要条件,对于X射线衍射,当光程差等于波长的整数倍时,晶面的散射线将相长加强。
2、扫描模式
(1)步进扫描
粉末衍射仪的一种工作方式(扫描方式)。试样每转动一步(固定的Δθ)就停下来,测量记录系统开始测量该位置上的衍射强度。强度的测量也有两种方式:定时计数方式和定数计时方式。然后试样再转过一步,再进行强度测量。如此一步步进行下去,完成指定角度范围内衍射图的扫描。
步进扫描一般耗费时间较多,因而须认真考虑其参数。选择步进宽度时需考虑两个因素:一是所用接收狭缝宽度,步进宽度至少不应大于狭缝宽度所对应的角度;二是所测衍射线线形的尖锐程度,步进宽度过大则会降低分辨率甚至掩盖衍射线剖面的细节。为此,步进宽度不应大于最尖锐峰的半高度宽的1/2,但是,也不宜使步进宽度过小。步进时间即每步停留的测量时间,若长一些,可减小计数统计误差,提高准确度与灵敏度,但将损失工作效率。
(2)定速连续扫描
是粉末衍射仪最常用的一种工作方式(扫描方式)。试样和接收狭缝以角速度比1:2的关系匀速转动。在转动过程中,检测器连续地测量X射线的散射强度,各晶面的衍射线依次被接收。计算机控制的衍射仪多数采用步进电机来驱动测角仪转动,因此实际上转动并不是严格连续的,而是一步一步地(每步0.0025°)跳跃式转动,在转动速度较慢时尤为明显。但是检测器及测量系统是连续工作的。连续扫描的优点是工作效率较高。例如以2θ每分钟转动4°的速度扫描,扫描范围从20~80°的衍射图15分钟即可完成,而且也有不错的分辨率、灵敏度和精确度,因而对大量的日常工作(一般是物相鉴定工作)是非常合适的。但在使用长图记录仪记录时,记录图会受到计数率表RC的影响,须适当地选择时间常数。
3、其他参数
测角仪:是粉末衍射仪上最精密的机械部件,用来精确测量衍射角。
发散狭缝:测角仪上用来限制发散光束的宽度。发散狭缝的宽度决定了入射X射线束在扫描平面上的发散角。
接收狭缝:测角仪上用来限制所接收的衍射光束的宽度。接收狭缝是为了限制待测角度位置附近区域之外的X射线进入检测器,它的宽度对衍射仪的分辨能力、线的强度以及峰高/背底比有着重要的影响作用。
防散射狭缝:测角仪上用来防止一些附加散射(如各狭缝光阑边缘的散射,光路上其它金属附件的散射)进入检测器,有助于减低背景。防散射狭缝是光路中的辅助狭缝,它能限制由于不同原因产生的附加散射进入检测器。例如光路中空气的散射、狭缝边缘的散射、样品框的散射等等。此狭缝如果选用得当,可以得到最低的背底,而衍射线强度的降低不超过2%。如果衍射线强度损失太多,则应改较宽的防散射狭缝。
Sollar狭缝:测角仪上一组平行薄金属片光阑,由一组平行等间距的、平面与射线源焦线垂直的金属薄片组成,用来限制X射线在测角仪轴向方向的发散,使X射线束可以近似的看作仅在扫描圆平面上发散的发散束。
4、检测过程中注意事项及可能遇到的问题分析
(1)同一份样品重现性差的可能原因:
①样品粉末颗粒大,导致样品中颗粒能产生衍射的晶面减少,使得某个峰很强,其他峰很弱。
解决办法:研磨或研磨过筛(研磨力度适宜,不可转晶,颗粒不可过细而破坏了晶体结构)
有些颗粒不大的样品,但是晶体的晶癖特征(主要是针状、棒状、片状:存在很窄的晶面),可能会导致择优取向,某些方向过度衍射,其他方向衍射不足,这时也可采用研磨的办法减少择优取向的影响。
②由于样品表面不平引起的误差。样品表面不平整,那么 X 射 线通过样品后就不能完全聚焦在聚焦圆上。从而使得峰变宽其不对称。
解决办法:在制备样品时,尽量使样品铺平整紧密。且样品表面应与 样品盘表面保持平行,样品不应过少或铺得过厚。
③样品本身在测试条件下不稳定:如样品易吸湿后转晶,常温下部分转晶等。
(2)同一个晶型,衍射峰强度不一样
不同批次结晶度存在差异,衍射峰强度变弱,结晶度变差。
结晶度:晶态和非晶态共存时结晶相所占比例,有时也指结晶完整度。
结晶完整的晶体:通常晶粒较大,衍射线强,峰尖锐且对称。
结晶度差的晶体:衍射峰宽而弥散。
(3)不同批次的样品,原本重叠的峰分开了,仍是同一个晶型
不同批次对应晶面的结晶情况不一样。比如,晶体在后期生长更完善后,结晶程度变高,峰变窄,进而两个峰能分开。
(4)同一个样品,衍射峰角度有偏移。
①所有角度均整体偏移,偏移角度在0.2度以内均正常,仪器测试误差,可通过软件调整。
②部分左偏移,部分右偏移,考虑是否发生转晶。
(5)晶型一样,晶癖不一定一样
晶型是内部结构,晶癖是外部表现。
同一个晶型,在不同的助(/反)溶剂的相互作用下,改变晶体生长动力学,促进或抑制某晶面生长,形成不同晶癖。
(6)对制剂检测:
①原料药为晶体,但载药量过低,低于仪器检出限。可以提高原料药载药量,采用相同的工艺制备制剂成品,确认原料药晶型在制剂工艺过程中是否发生转晶。
②原料药是晶体,但在制剂工艺生发生转晶,转为无定型,那么需要确定制剂中原料药载药量高于检测限。
二、差示扫描量热法(DSC)
1、测量原理:
在对供试品与热惰性的参比物进行同时加热(或冷却)的条件下,当供试品发生某种物理或化学的变化时,将使热效应改变,供试品和参比物质之间将产生温度差(△T)。这种在程序控制温度下,测定供试品与参比物之间温度差与温度或时间)关系的方法称为差热法(DTA)。而测量输给供试品与参比物热量差(dQ/dT)与温度(或时间)关系的方法称差示扫描量热法(DSC)。
2、DSC图谱分析
(1)玻璃转化温度(Tg):
非晶聚物有三种力学状态,它们是玻璃态、高弹态和粘流态。在温度较低时,材料为刚性固体状,与玻璃相似,在外力作用下只会发生非常小的形变,此状态即为玻璃态:当温度继续升高到一定范围后,材料的形变明显地增加,并在随后的一定温度区间形变相对稳定,此状态即为高弹态,温度继续升高形变量又逐渐增大,材料逐渐变成粘性的流体,此时形变不可能恢复,此状态即为粘流态。我们通常把玻璃态与高弹态之间的转变,称为玻璃化转变,它所对应的转变温度即是玻璃化转变温度。
玻璃化转变在曲线中具体表现为基线的整体提升。
(2)熔化峰:
熔融峰可显示熔融发生时的温度(oneset值),峰值温度(peak值),但这两种温度值与熔点值可能并不一致(如受升温速率等影响);
物体的熔化是一个过程,包括初熔点、融程和终熔点。
① 外推起始温度Tei ℃,即“初熔点”:外推基线与对应于转变开始的曲线最大斜处所作切线的交点所对应的温度。
② 峰温度Tp ℃,即“终熔点”:峰达到的最大值(或最小值)所对应的温度。终熔点,我们认为是样品刚刚好完全融化。
③ 外推终止温度Tef ℃:外推基线与对应于转变结束的曲线最大斜处所作切线的交点所对应的温度。
目前,我们测试初熔点Tei数据稳定,但是终熔点Tp受实验样品量、升温速率影响,数据有微小差异。
一般都是对于小分子样品以DSC 峰基线与峰切线的交点(Onset Point)作为熔点。
3、检测过程重注意事项
(1)快速升温,DSC峰型变大(相当于灵敏度提高),特征峰向高温偏移,相邻峰分离能力下降;慢速升温,相邻峰分离增加,峰型较小。常用10K/min
(2)样品量小,特征温度较低,更“真实”
相邻峰分离能力增加,峰型较小;
样品量大,增加峰响应,峰型变宽,特征峰向高温偏移。--这是因为较多的供试样品在坩塌中厚度较大,这种情况使供试样品内的传热速度减慢,从而形成较大的温度梯度,导致供试样品的吸热峰峰宽变大,既降低了供试样品吸热峰的分辨率,又使供试样品的熔点和熔距的测定值产生偏差。
(3)样品处理:
对于不同条件的DSC样品,如何选择合适的制样方式?
A.块状样品:建议切成薄片或碎粒;
B.粉末样品:使其在坩埚底部铺平成一薄层;
C.堆积方式:一般建议堆积紧密,有利于样品内部的热传导;对于有大量气体产物生成的反应,可适当疏松堆积。
D.样品粒度越小,放热峰越大越尖锐。粒度过大或者颗粒形状不规则,可能会导致熔距变大。可研磨(研磨力度不可过大,破坏晶体构象),紧密堆积。
E.选择能与样品兼容的材质坩埚
三、热重分析(TGA)
TGA检测原理、操作及分析比较简单,所以简单描述一下其原理及检测过程中的注意事项。
1、原理
热法(TG)是在程序控制温度下,测量物质的重量与温度关系的一种方法。记录的重量变化与温度或时间的关系曲线即热重曲线(TG曲线)。由于物相变化(如失去结晶水、结晶溶剂、转晶或热分解等)时的温度保持不变,所以热重曲线通常呈台阶状,重量基本不变的区段称平台,利用这种特性,可以区分样品中所含的水分是吸附水(或吸附溶剂)还是结晶水(或结晶溶剂),并根据平台之间的失重率可计算出结晶水或结晶溶剂的比例。
2、影响TGA数据的因素
(1)样品量过多,导致传热和挥发物挥发速度变慢,导致相邻失重转变靠近,因此,在灵敏度许可的范围内,样品量尽可能少。
(2)样品的细度、装填紧密程度对传热及挥发物的挥发有影响,一般要求样品细致均匀,装填成均匀的薄层(相对紧实)。
(3)气体的浮力和对流
浮力的影响:样品周围的气体因温度的升高而膨胀,比重减小,则样品的TGA值增加。
对流的影响:对流的产生使得测量出现起伏。
(4)挥发物的再凝聚
凝聚物的影响:物质分解产生的挥发物质可能凝聚在与称重皿相连而又较冷的部位上,影响失重的测定结果。
(5)样品与称量皿的反应
反应的影响:某些物质在高温下会与称量皿发生化学反应而影响测定结果。
(6)升温速率的影响
升温速率的影响:升温速率太快,TGA曲线会向高温移动;速度太慢,实验效率降低。
总结:晶型的检测表征方法有多种,在本文中仅简单描述了三种比较常用的检测方法及其注意事项,检验操作比较简单,但需要对结果进行有效的判断。鉴于笔者水平,若有不对之处,希望大家指出。










加载更多