几种蛋白质组学技术原理介绍及比较
蛋白质组学
原理介绍及比较
过去的几十年,蛋白质组学研究技术不断发展、完善,已形成了多种广泛应用的技术手段。它们特点不同、优势不同、适用侧重面也具有一定差异。那么,如何选择适合的蛋白质组学技术以适用自己的课题研究呢?小析姐分享几种蛋白质组学研究策略,并对其进行了比较。
蛋白质组学研究可以从整体水平观察细胞内蛋白质的组成及其变化规律,整体性、概括性地了解生命体在某一生理状态下的蛋白表达、修饰、相互作用等,从而揭示蛋白功能,为生命科学的理论研究提供重要的参考意义。蛋白质组学具有广泛的应用范围,能用以研究生理、病理、药物作用机制;用于找寻疾病诊断的Biomarker、筛查适应性的治疗药物等临床诊断的研究;也可以用作精准医疗如药物开发、个性化治疗等方面的研究。
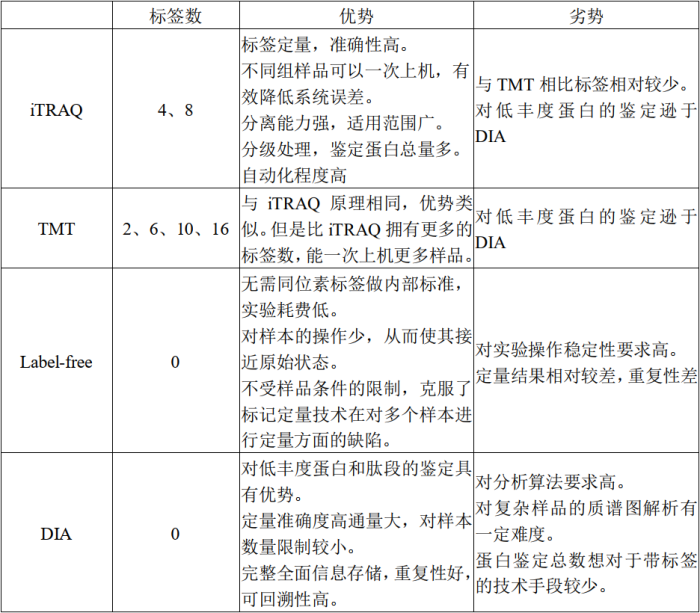
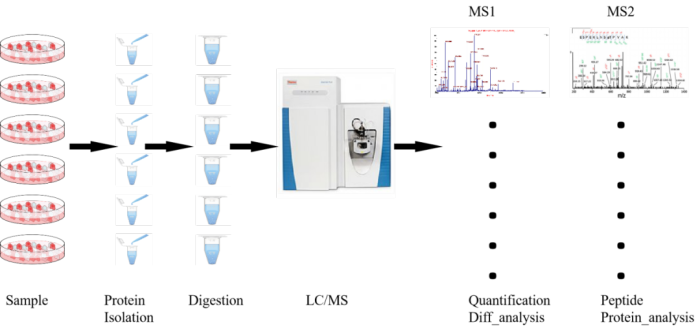
现阶段市面上流行的、商业化应用较广的蛋白质组学解决方案共有4中:TMT、iTRAQ、DIA和Label-free。其中TMT和iTRAQ具有相似原理,都是通过标签实现样本的区分以及蛋白的定量,可一次上机多个样本进行分析、比较,准确度较高并能有效降低系统误差。DIA和Label-free均不带标签,利用具有一定区分度、代表性的肽段实现肽段的定量。定量结果准确度相对较差,而且只能实行单样本上机,受系统误差影响相对较大。
TMT(Tandem Mass Tag)技术由美国Thermo Fisher Scientific开发的一种多肽体外标记技术。该技术采用了6标(TMTsixplex™ Isobaric Label Reagent Set)、10标(TMT10plex™ Isobaric Label Reagent Set )、16标(TMTpro 16plex)同位素标签,通过与肽段特异氨基酸位点相连实现不同来源的肽段标记,然后进行串联质谱分析,监测碎裂下来的标签实现肽段定量。一次实验可灵活比较最多16种不同样本中蛋白质的相对含量。
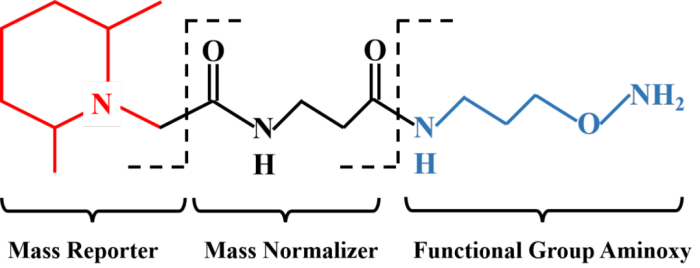
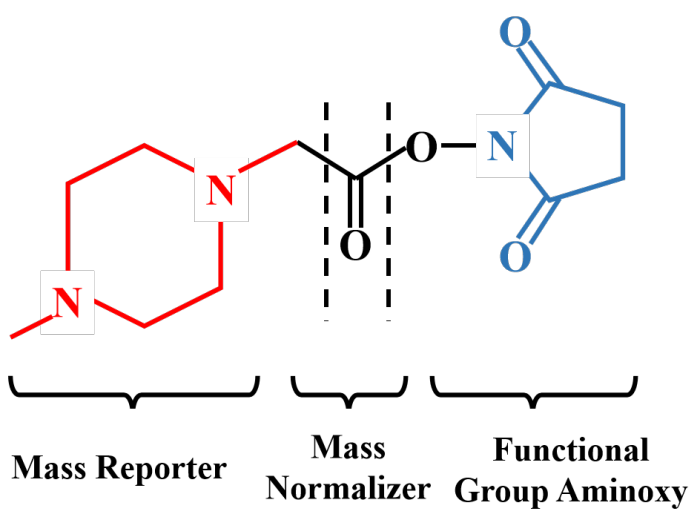
TMT实验中,经过蛋白质提取获得组织细胞中的全蛋白,全蛋白在胰酶作用下被酶解成肽段,TMT试剂对所获得的全部肽段进行标记。TMT试剂主要由3部分组成:报告基团、平衡基团和肽反应基团。以6标试剂为例:报告基团相对分子质量分别为126、127、128、129、130和131;平衡基团相对分子质量分别为103、102、101、100、99和98,这样就形成了6种相对分子质量均为229的等量异位标签。
TMT标签试剂通过肽反应基团与肽段上赖氨酸及N端氨基酸残基相连。被标记肽段经过色谱分离后直接进入串联质谱仪中进行定性及定量检测。一级质谱中,任何一种TMT试剂标记的不同样品中的同一肽段表现出相同的质荷比;二级质谱中,报告基团、平衡基团、连接基团之间的化学键断裂释放出TMT报告基团和平衡基团。平衡基团发生中性丢失,报告基团被质谱仪检测并记录,在质谱低质量区产生了10个TMT报告离子峰,其强度反应了该肽段在不同样品中的相对表达量信息,另外二级质谱中的肽段碎片离子峰质荷比反应了该肽段的序列信息;这些质谱原始数据经过数据库检索,可得到蛋白质的定性和相对定量信息。
iTRAQ(isobaric tags for relative and absolute quantitation)与TMT原理相似,都是采用不同标签对不同来源的蛋白进行标记并利用质谱检测的标签强度对含量进行相对定量。不同的是标签的类型及数量。iTRAQ最多只有10标,也就是说采用iTRAQ策略的蛋白质组学研究,一次最多只能上机10个样本,通量相对受限。而TMT最多可达16标,相对而言具有更高的通量。
Label-free和DIA采用非标签标记定量策略。Label-free通过比较质谱分析次数或质谱峰强度,分析不同来源样品蛋白的数量变化。该方法认为肽段在质谱中被捕获检测的频率与其在混合物中的丰度成正相关。因此,蛋白质被质谱检测的计数反映了蛋白质的丰度,通过适当的数学公式可以将质谱检测计数与蛋白质的量联系起来,从而对蛋白质进行定量。
按照其原理主要分为两种:
第一种spectrumcounts类的非标记方法,发展比较早,已经形成多种定量算法。
其主要的原理是以MS2的鉴定结果为定量基础,各种方法的差别在于后期算法在大规模数据上的修正。
第二种非标记定量的原理是以MS1为基础,计算每个肽段的信号强度在LC-MS上的积分(如Maxquant)。
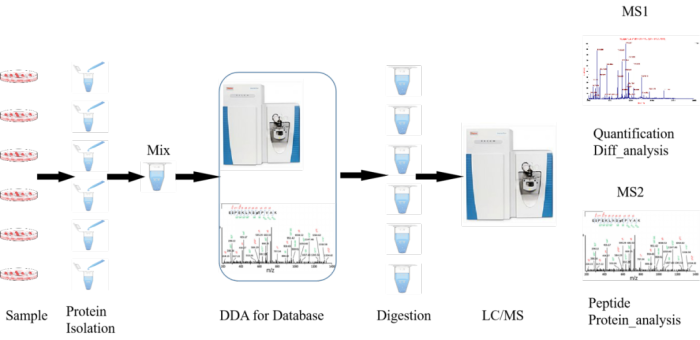
DIA技术融合了传统蛋白质组学“鸟枪法”(shotgun)和质谱绝对定量“金标准”选择反应监测/多反应监测(SRM/MRM)技术的优势和特点。主要采用数据非依赖采集模式(Data-independentAcquisition,DIA),结合传统的数据依赖采集模式(Data-dependentAcquisition,DDA)构建参考谱图库,通过液质联用技术(LC-MS/MS)将质谱整个全扫描范围分为若干个窗口,高速、循环地对每个窗口中的所有离子进行选择、碎裂、检测,从而无遗漏、无差异地获得样本中所有离子的全部碎片信息。将得到的数据利用主流分析软件Spectronaut,进行蛋白组学的鉴定、定量及差异蛋白分析等。具有全景式扫描、数据可回溯等优势。
上述蛋白质组学研究服务解决方案是当下相对来说应用最为广泛的几种方案。这几种研究策略具有不同特点。
1、分离能力强:可分离出酸/碱性蛋白,小于10KD或大于200KD的蛋白、难溶性蛋白等;
2、适用范围广:可以对任何类型的蛋白质进行鉴定,包括膜蛋白、核蛋白和胞外蛋白等;
3、高通量:可同时对多个样本进行分析,特别适用于采用多种处理方式或来自多个处理时间的样本的差异蛋白分析;
4、结果可靠:定性与定量同步进行,同时给出每一个组分的相对表达水平、分子量和丰富的结构信息;
5、自动化程度高:液质联用,自动化操作,分析速度快,分离效果好。
1、重复性好:DIA数据完整性为98%,DDA数据完整性为49%;
3、定量准确性高,定量准确度与MRM技术相当,CV值可达10%左右;
6、样本量大:DIA技术可以在一次实验中同时对几十、上百例样本进行差异表达蛋白质进行分析。
最后,小析姐分享几种蛋白质组学研究策略,并对其进行了比较。










加载更多