【干货】浅析ICP-MS法测定药品中元素杂质的方法学要求
国际人用药物注册技术协调会议(ICH)Q3D元素杂质指南对元素杂质的分类、制剂日允许暴露量与浓度限度之间的转换等做了详细阐述,有利于帮助药企通过风险评估来决定对哪些元素进行额外控制,为药品制订合理的元素限度[1]。2017年6月中国获得批准正式加入ICH,这意味着中国的药品监管也将逐步转化和实施国际ICH 的标准和指南,同时提高了我国各类化学药品申报的标准。《中国药典》2020版通则增修自2018年8月起已经发布了十五批修订公示,2018年12月28日发布的第三批公示包含了元素杂质限度和测定指导原则(第一次征求意见稿)[2],表明我国也将开始实施药品中元素杂质控制。
近年来发展迅速的电感耦合等离子体质谱分析技术(ICP-MS)是元素分析领域非常先进的技术,既可以定量测定元素含量,也可以定性分析元素种类,具有极低的检出限,其可检测的浓度能低至ng·kg-1级,较高的灵敏度和精密度、线性范围宽、抗干扰能力强,并且能提供精确的同位素信息的分析特征等优点,已经广泛应用于药品行业中元素杂质控制[3-7]。
方法学验证是对测定方法的评价,是建立新方法的研究内容和依据。在药品研发过程中需要根据产品的需求不断调整和优化检测方法,并做出完整的方法学验证,证明该方法可以用于药品的质量控制。本研究结合现行版的美国药典、欧洲药典、《中国药典》相关通则及相关文献,探讨并总结相对全面、科学的ICP-MS分析药品中元素杂质的方法要求。
如表1所示,美国药典、欧洲药典、《中国药典》通则中均建立了ICP-MS仪器方法要求(USP40<730>Plasma Spectrochemistry[8]、EP9.2<2.2.58>Inductively Coupled Plasma-mass Spectrometry[9],ChP2020<0412 >电感耦合等离子质谱法[10])。
表1 各国药典通则ICP-MS仪器方法要求汇总
验证内容 |
USP<730> |
EP <2.2.58> |
ChP<0412 > |
系统适用性 |
/ |
1、仪器调谐,记录灵敏度,短期和长期稳定性,优化仪器参数。 2、对分辨率和质量轴进行调谐。 3、评估等离子体分解氧化物的效率,使干扰最小化,Ce/CeO或Ba/BaO的比例<3%;用Ba和Ce减少双电荷离子的形成,双电荷离子的信号与指定元素的比值<2%。 4、通过在样品序列的最后运行标准品来检查长期稳定性,控制锥体上的盐沉积物是否在整个运行期间降低了信号。 |
/ |
线性与范围 |
类别1:居中100.0%浓度的验证范围为80.0%~120.0%,相关系数r≥0.995; 类别2:验证范围为70.0%~130.0%,相关系数r≥0.99。 |
相关系数r≥0.99 |
相关系数r≥0.99 |
专属性 |
方法应该能够确保在样本中,每个元素都能被明确的检测出。 |
/ |
/ |
检测限 |
/ |
/ |
不少于7份空白样品溶液,连续测定空白样品溶液响应值的3SD对应的待测元素浓度。 |
定量限 |
定量限QL=10份空白溶液的标准偏差×10。应至少能够定量准确分析至限度的50%。 |
定量限度低于限度值。 |
不少于7份空白样品溶液,连续测定空白样品溶液响应值的10SD对应的待测元素浓度。 |
准确度 |
用标准溶液配制含50%~150%限度浓度目标元素的标准溶液。 类别1:平均回收率在95.0~105.0%之间; 类别2:每个杂质的三份不同浓度的元素回收率均在70%~150%之间。 |
含量测定:回收率范围90%~110.0%; 微量元素测定:回收率范围80%~120.0%。 |
/ |
重复性 |
样品溶液:6份样品(相同批次),分别按限度加入标准元素杂质。 类别1:RSD应不超过5%; 类别2:每种元素杂质的RSD应不超过20%。 |
含量测定:RSD%≤3%; 杂质测定:RSD%≤5%。 |
/ |
中间精密度 |
不同的实验员,在不同的时间或用不同的仪器,按重复性测定(应至少包括3个不同因素)。类别1:RSD应不超过8%; 类别2:每种元素杂质的RSD应不超过25%。 |
/ |
/ |
注:类别1:含量;类别2:定量杂质及限度测定。
此外,如表2所示,美国药典、欧洲药典通则中还单独建立了药品中元素杂质残留检测方法要求(USP40<233>Elemental Impurities-Procedures[11],EP 9.2<2.4.20>Determination of Metal Catalyst or Metal Reagent Residues[12]),目前中国药典还没有单独建立药品中元素杂质残留检测方法要求,可以借鉴欧美药典通则增订适用于我国具体情况的相关通则。
表2 各国药典通则元素杂质残留检测方法要求汇总
验证内容 |
USP<233> |
EP <2.4.20> |
系统适用性 |
待测物漂移不大于20%。 |
分析当天必须进行系统适用性测试,以确保样品制备和测量系统合适。 |
线性与范围 |
满足准确度要求。 |
符合回收率要求。 |
专属性 |
分析方法应当能够明确检测每种目标元素(见USP<1225>Validation of Compendial Procedures[13]),共存的其他成分(包括其他目标元素、基质成分)不干扰检测。 |
分析方法应当能够明确检测每种目标元素,共存的其他成分(包括其他目标元素、基质成分)不干扰检测。 |
检测限 |
/ |
信号强度明显区别于空白溶液,检测限不超过限度值的0.5倍。 |
定量限 |
满足准确度要求。 |
定量限度低于限度值。 |
准确度 |
三个不同浓度水平下加样回收率平均值:70.0%~150.0% |
三个不同浓度水平下加样回收率:70.0%~150.0%。 |
重复性 |
6个加标样品或3个浓度水平九份样品RSD%≤20%。 |
/ |
中间精密度 |
不同仪器、不同分析人员、不同时间或其组合,与重复性共12次测定结果RSD%≤25%。 |
/ |
本次研究结合各国药典相关通则及相关文献,分别从系统适用性、线性与范围、专属性、检测线与定量限、准确度、精密度等方法要求对ICP-MS分析药品中元素杂质方法进行讨论,供行业内参考。
1、系统适用性
EP<2.2.58>提出了对ICP-MS仪器分析较全面的系统适用性要求,包括仪器调谐要求及序列运行稳定性要求。仪器调谐要求包括仪器分辨率及质量轴的调谐,并明确了调谐后灵敏度、双电核、氧化物的要求。序列运行稳定性要求通过在样品序列的最后运行标准品来检查长期稳定性,控制锥体上的盐沉积物是否在整个运行期间降低了信号。USP<233>提出比较样品序列运行过程中分析样品测定前后150%限度浓度对照品结果,测定元素杂质信号的漂移,以控制整个运行期间系统的稳定性。根据各国药典的要求,总结ICP-MS元素杂质分析方法较全面的系统适用性要求为仪器开机后用调谐液调谐,配置好仪器参数,记录仪器性能需符合药典要求,样品序列运行过程中分别取150%限度浓度对照品溶液进样分析,信号漂移不得超过20%。
2、 专属性
USP<730>、USP<233>、EP<2.4.20>规定分析方法应当能够明确检测每个目标元素,共存的其他成分不干扰检测,例如其他目标元素、基质成分等。上述药典均未明确ICP-MS元素杂质分析方法专属性的具体做法。
ICP-MS分析过程中受到的干扰主要包括同质异位素、多原子离子、双电荷离子、物理干扰、基体效应、记忆效应等[10]。通过调研ICP-MS测定元素杂质分析方法的文献[14-17],发现在方法建立或验证ICP-MS方法时进行专属性相关研究工作的较少,仅有少数研究者针对专属性进行了针对性的研究。
成勇在微波消解-ICP-MS法测定二氧化钛中痕量元素[18]研究中,系统地研究了H2SO4-HCl和HNO3-HF两种消解消解体系,证明H2SO4-HCl消解体系对待测目标元素产生较严重的多原子质谱干扰,HNO3-HF消解体系不容易形成多原子干扰,不影响待测目标元素测定;对基体元素Ti与消解试剂、Ar、O、H等ICP-MS中的主要元素结合所形成的多原子离子等质谱干扰进行了试验,研究出了钛基多原子离子存在的主要潜在干扰,据此优选不受其干扰的各待测元素的分析同位素,保证了方法的专属性。
DianePaskiet等探索三种不同pH萃取剂从弹性体胶塞中提取元素杂质,以评估包材浸出至药品中元素杂质的风险 [19],研究过程中发现,由于萃取剂中含有盐酸,待测目标元素75As会受到40Ca35Cl和40Ar35Cl的多原子离子干扰。通过向ICP-MS仪器反应池中引入氧气,75As与16O可反应生成质量数为91的75As16O,40Ca35Cl和40Ar35Cl不与氧气反应,并保持质量数为75,因此改为测定质量数为91的75As16O代替As元素的测定,从而减少了多原子对待测目标元素的干扰。
笔者针对ICP-MS分析过程中可能受到的干扰,对方法验证过程中专属性内容进行了探讨,可供同行业参考。
(1)各待测元素均应有各自的m/z,且待测元素的同位素之间不互相干扰检测;若待测元素的同位素之间有重合,应使用仪器校正方程自动修正。开发方法时根据样品基质含有的元素,考察可能的多原子干扰目标待测元素情况。
(2)空白溶液中待测元素的响应值,干扰应远低于限度浓度;
(3)做外标法时,对照品和样品基质如有差异,可能导致离子化效率不一致,应考察基质干扰对测定的影响,可采用以供试品溶液作为基质,在基质中加入限度浓度对照品,与相应浓度对照品进行比较,按下式方法计算基质干扰,待测元素基质抑制或增强均在一定范围内,说明基质干扰较小,通过上述几方面工作,可以证明专属性良好。
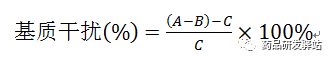
式中:A为加标供试品溶液中各元素杂质响应值(cps);B为供试品溶液中各元素杂质响应值(cps);C为限度浓度对照品溶液中各元素杂质响应值(cps)。
3、线性与范围
结合各国药典要求,对于低浓度,特别是微量元素分析,并且浓度范围不大,检测器响应可靠,背景干扰较小,一般有3个浓度点(包括2点标准溶液及空白)即可。对于平时测量的样品浓度范围较宽,并且检测器响应不完全是一次曲线(直线)的,可以采用二次曲线或不分段校正方式,以减少数据偏差,数据点可以选择4~6个。范围应包括样品的最高、最低浓度且具满足精密度、准确度、线性的要求,可以设计为等比或等差浓度点。标准曲线应该采用合适的统计学方法,例如最小二乘回归,各国药典要求均为线性相关系数应大于0.99。USP<730>对标准溶液的稳定性做出了要求,用于微量分析的标准溶液的有效期是有期限的。一般取决于浓度、存储容器的类型、存储条件。由于这些原因,浓度小于10ppm(W/V)的标准溶液应保留不得过24小时,除非另有验证数据证明其稳定性。
4、检测限与定量限
检测限(LOD)是分析方法最重要参数之一,也是争议较大的参数之一。调研国内文献[20-21]时,发现多数研究者对于空白样品、测定次数及根据所需置信度选择的系数要求不是很明确,笔者对LOD、定量限(LOQ)定义、计算公式、具体做法等来源进行了调研[22]。
国际纯粹与应用化学联合会(IUPAC)将检出限定义为某特定方法在给定的置信度区间内能够合理地检测出的最小分析信号求得的最低浓度[23]。检出限的公式表示为:LOD = (kσB )/ S,其中k表示根据所需置信度选择的系数,σB表示空白的标准偏差,S是该分析物的校准曲线的斜率(即灵敏度)。Kaiser[24]认为k取值3较为合适,因为这代表大多数应用中计算的检测限的95%置信区间,因此3是该计算中最常用的值。
AMC(Analytical Method Committee分析方法委员会)推荐计算LOD时空白测定应不少于10次,建议使用真实空白,但在实际分析中很难得到,许多分析工作者使用试剂空白或接近空白代替。IUPAC建议σB应通过实验以足够多的测定次数求出, 如20 次;环境保护署(EPA)建议根据对目标分析物空白样品进行7次重复分析,计算“方法检出限”,其浓度为计算出的仪器检测限浓度的2或3倍[25]。Chp<0412>规定LOQ为不少于7份空白样品溶液,连续测定空白样品溶液响应值的3倍标准偏差对应的待测元素浓度。
LOQ通常与LOD一起计算。LOQ是对LOD的保守估计,有时用作方法的报告限度[26]。对于每种分析物,LOQ代表可以足够准确地测量和报告的最低浓度。可以任意定义精密度和准确度的理想水平,因此没有用于计算LOQ的具体公式。但是,所需的精密度水平通常约为10%(报告为相对标准偏差),这意味着使用空白标准偏差的10倍来计算LOQ[27]。根据定义,LOQ不能低于LOD,并且不应等于LOD。
5、准确度
USP <730>、USP<233>、EP<2.4.20>、EP<2.2.58>规定在样品消化或溶解之前,加入适量标准物质制备3个不同浓度的溶液计算加样回收率。根据欧美药典要求,建议取9份供试品溶液,按50%、100%、150% 限度浓度分别加入标准物质,作为低、中、高3个不同浓度的加标供试品溶液,每个浓度制备3个平行样品,进行加样回收试验,验证方法准确度。
(2)中间精密度

展源
何发
相关文章
-

ICP-MS 法法测定水质中的重金属元素
2020-05-27
-

AAS法分析茶叶中的铅,镉,砷
2020-05-27
-

定量分析中怎样选择内标法或外标法
2020-05-27
-

色谱定量分析中怎样选择内标法或外标法
2020-05-27
-

色谱定量分析中 选内标法or外标法?
2020-05-27
-

QuChERS-HPLC法检测奶粉中双氰胺
2020-05-27
-

HPLC法检测VE中苯并芘
2020-05-27
-

检测有机氯类农药,气相色谱法检测法
2021-01-12
-

高效液相色谱法分析人参渣中人参多糖的含量
2020-09-11

加载更多