保留时间为何不恒定
“液相故障与排除”是分析工作者经常遇到的问题,而这两者都与液相色谱方法中的保留时间变化有关,本文就这方面进行了一些探讨。
流动相组成的敏感度
流动相的组成对保留时间的影响是十分灵敏的,例如在使用反相液相色谱法分离两个分子量约为400Da,不含离子化基团的异构体,流动相为乙腈和水。通常这两个异构体的保留时间相隔约1min而被分开,分离度大于2。在等度洗脱的条件下,如果乙腈的比例有1%的变化,会导致保留时间2min的漂移。若采用一个较缓慢的梯度(超过10minB变化5%)时,在初始及梯度结束时1%乙腈的改变会导致保留时间会有1min的漂移。显而易见,不论以上哪种方法,流动相组成的一个小小误差就足以使保留时间发生变化,进而使只采用保留时间对色谱峰的定性鉴别得出错误判断。
相似的问题
在我的实验室中遇到过类似的问题,所以很值得回忆一下问题本身及解决方法。在分离分子量1000 Da 的多肽时,我们采用反相色谱柱250mm×4.6 mm,5μm,流速1.5ml/min,柱温35℃,流动相为乙腈及水(含0.1%三氟乙酸)梯度洗脱,19%~24%的乙腈运行30min。 即使用新的泵(新的单相阀和新的泵垫),保留时间在超过连续3次进样后变化了大约1min,如图1a所示。
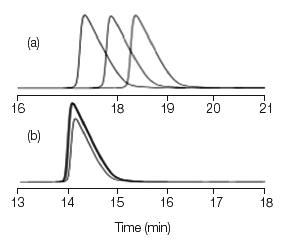
图1. 多肽样品在线混合时保留时间的变化。
(a) A= 水(0.1%三氟乙酸),B=乙腈(0.1%三氟乙酸);
(b) A=10%乙腈,90%水(0.1%三氟乙酸),B=30%乙腈,70%水(0.1%三氟乙酸)。
问题的根源与液相色谱仪系统的溶剂在线混合能力有关,注意上述方法中的流动相梯度变化是一个很小的范围,在超过30min的时间变化5%,或者≈0.17%/min。大多数液相色谱泵的制造商规定准确度比例为±0.5%~1%/min。显然我们所想要的流动相梯度控制要求要高于制造商所规定的指标。
解决方法:预混合
解决这一问题的方法很简单,我们采用事先混合好的流动相,A为10%的乙腈,90%含0.1%的三氟乙酸的水;B为30%的乙腈和70%含0.1%的三氟乙酸的水。梯度控制为在30min,流动相B变化范围为40%~65%,等同于乙腈-水(0.1%三氟乙酸)18%~23%的变化。在此条件下,连续进样3次保留时间的变化<0.1 min(参见图1b)。从上述液相系统可见,梯度变化是在30min25%,或者≈0.8%/min,将梯度的斜率降低了5倍,显然,该系统可以在这个水平上产生很好的效果。
我们可以使流动相A含15%的乙腈,而流动相B含有25%的乙腈,并且相应调节梯度程序,会得到更好的结果。为什么不采用流动相A含19%的乙腈,流动相B 含24%的乙腈,梯度程序从0~100%呢?当保留时间对流动相组成比例的微小变化非常敏感的时候,手动溶剂混合的正常误差会变得非常大而造成方法的失败。所以,选择A 及B溶剂的浓度在所期望的范围之外,如果需要调节保留时间至所希望的数值,可以有空间将初始及最终设定“拧”在一起。例如,由于A或B配制的误差,我们可以调节梯度由30min41%~64%的流动相B代替 40%~65%的B,恰好留下了一个小小安全余地。
我的推测是预混合的技巧可以解决在最初碰到的保留时间变化的问题。因A流动相组成1%的变化导致保留时间1min的变化可能是过分的,但是预混合的确可以减少这一问题。所以将A中加入5%的乙腈及B中15%的乙腈代替水(A)及乙腈(B)。现在设定控制器以±0.5%~1.0%的准确度释放10%B,梯度程序将可以设定为50%B并且有效的准确度可被提高到±0.05%~0.1%。操作者可在规定的限度内以0.1%的增量调节浓度程序直到获得正确的保留时间为止。类似的对策可以用于较缓梯度程序的同样样品。
对于等度或梯度程序方法,即使条件与上述要求不同,预混合都有着另外的优势。对于我们分离多肽的方法,我们观察到预混合可以减小基线噪音,如图2所示。比较两条基线,相对于流动相比率的变化,预混合后的基线噪声大约减低了5倍。最初方法的流动相A为水(三氟乙酸),B为乙腈(三氟乙酸)[100%的范围,图 2(a)],相反的条件流动相A为10%乙腈,B为30%的乙腈[20%的范围,图2(b)],在变化幅度上减小了5倍。
采用流动相预混合的方法减小基线噪声是非常容易看到且不是一个最近发现的现象。对很多方法来说,甚至在A中预混合5%B溶剂,在B中混合5%的A溶剂将会产生有益的结果。

图 2. 多肽样品梯度洗脱在线混合时的基线。
(a) A= 水(0.1%三氟乙酸),B=乙腈(0.1%三氟乙酸);
(b) A=10%乙腈,90%水(0.1%三氟乙酸),B=30%乙腈,70%水(0.1%三氟乙酸)。
最初的漂移
第二个问题是批分析最先进样的前几针的保留时间会有漂移并逐渐趋于一恒定值,通常保留时间漂移经过15次进样后趋于恒定值,这也是分析工作中时常发生的问题之一。
硅胶表面
问题缘于固定相,在反相液相中作为载体来键合固定相的硅胶颗粒为无定型硅及其氧化物的聚合物,它的末端为硅羟基(-Si-OH)基团,裸露在硅胶颗粒的表面,我们试想一下这些游离的硅羟基,如图3(a)所示,Kirkland及其他人的工作显示硅胶表面十分复杂,例如,硅羟基可以偕二羟基存在,见图3(b),若空间合适的话,两个相邻的硅羟基以-H相连形成复合的硅羟基基团,见图3(c),游离的硅羟基的酸性强于偕二羟基和复合的硅羟基基团。
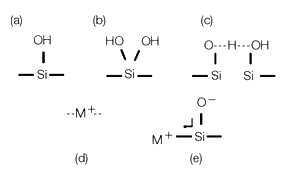
图3. 硅胶表面:(a)游离的硅羟基,(b)硅偕二羟基,(c)复合硅羟基,(d)嵌入的金属,(e)活化的硅羟基。
早些时候,A型硅胶含有明显数量的金属,如铝和铁,见图3(d),对酸性样品来说,提供了离子交换的场所,并且,若位置合适的话,会从相邻的硅羟基基团上夺去电子而形成离子化的或“活化”的硅羟基见图3(e)。现在通用的高纯度硅胶也称B型硅胶,其大大降低了金属的含量及酸性的游离硅羟基,因此硅胶表面是大量的硅偕二羟基和复合的硅羟基基团,见图3(b)和图3(c),这使得硅胶表面更少可能造成碱性化合物的拖尾。
键合固定相通过硅烷基醚键与硅胶表面相连(-Si-O-Si-R, 其中 R 为C18 或其它键合功能基团)。键合的固定相覆盖了约半数的可利用的硅羟基,因此,可简单描述为固定相表面可能含有50%的键合相和50%的未键合的硅羟基基团。似乎这样描述硅胶表面过于简单,其它类型的相互作用也可能存在。
各种不同的硅胶表面特性(键合相、游离硅羟基、金属等等)有着不一样的容量和平衡速率,而这些都将影响色谱的行为。很多色谱工作者会熟悉老的A型硅胶, 酸性的硅羟基对碱性化合物造成很糟糕的峰拖尾现象,这是由于碱性基团和离子化或活化的硅羟基位点相互作用的结果,见图3(e)。在这种情形下,一种广泛用于减少拖尾的办法是在流动相中加入三乙胺(例如,25mM)“淹没”这些离子基团的位点,可以改善峰形。这对于高纯度的B型硅胶则没有太大的必要,但是这些特性仍以不同的容量及平衡存在于硅胶表面。
该怎么办?
复杂的固定相表面的最终结果是对于一个给定的样品来说,每一种基团不会有一样的平衡速率,有时有必要多次进样直到这种缓慢的平衡位点被样品所饱和,并且新的样品进样时不再改变。这个过程就是通常所说的好像“灌满”或“粘住”色谱柱。尽管可以用样品或对照品进样来平衡色谱柱直到保留时间稳定,但更快捷的方法是用高浓度的样品溶液进样以达到同样的结果。通常总样品量是重要的,而不是接触的时间。所以,你可以接连用几个高浓度的样品溶液进样,然后等待方法的运行,而不是采用几个单独的运行。
“灌注”需要因色谱柱表面和样品会有很大的变化,所以,通常最好是用经验的方法来进行,在谈到过的制备分离的例子中,显示要用15次进样才可达到充分的柱平衡,我会试试用10倍浓度的样品进样2次减短这一过程,看是否会有同样的效果。
即使在现在的情形下,“灌注”的需要不那么明显,当一个方法刚开始运行通常还是会有很多改变发生,所以,最后弃用第一、二针。例如,在我接触过的一个实验室,大多数方法建议在首次系统适用性运行之前,以标准曲线的高浓度2~3次进样。
总结
本期讨论的2个例子在解决方法上有一个共同的主题,如果你对液相色谱仪的特性及分离过程有很好的了解,则可以采用适当的调节以获得很好的结果。
这使我想起我的一个朋友说过的话,他总是告诉他公司的员工,“记住5个P”——“ Prior Planning Prevents Poor Performance”,意思是事先的计划可避免不好的结果,即防患于未然。尽管他的生意只是为农场和牛奶场供应饲料,但5个“P”对于我们在分析化学工作的人来说也具有很大的意义。
《实验与分析》
展源
何发
相关文章
-

气相色谱法之保留时间
2021-09-21
-
色谱保留时间为何漂移?
2023-06-29
-

保留时间为何不恒定?
2020-05-27
-

保留时间为什么重现性差?
2023-09-26
-

HPLC常见问题:保留时间漂移!
2022-08-22
-

保留时间漂移 该如何是好?
2020-05-27
-

保留时间不稳定漂移原因有很多!
2024-03-20
-

气相色谱法之保留时间,知多少?
2023-02-21
-

保留时间不重现怎么办?3招搞定!
2023-04-04

加载更多