卡马西平的转变热力学研究分析
卡马西平的转变热力学研究
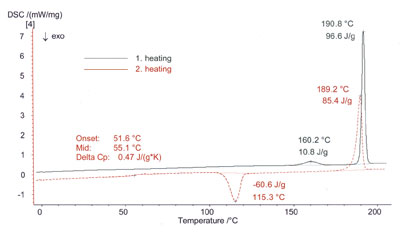
图1. 卡马西平,晶型III,连续熔融,1.23mg,铝坩埚,带孔盖;升温:-40~205℃/10K min-1;降温:205~ -40℃/40K min-1;升温:-40~205℃/10K min-1。
随着对药品的质量要求越来越高,热分析已成为控制药品质量、新药研究及新剂型开发的重要检测方法之一,美国、英国、日本和中国药典均收载了热分析方法。差示扫描量热法(DSC)是最常用的热分析方法,在药物熔点、纯度、多晶型的测定、药品溶剂化物和水份的测定、药品的相容性和稳定性测定等方面具有广泛的应用。
药物的多晶态是普遍存在的现象,即相同化学成分的物质存在多种不同的晶态,这些晶态中只有部分晶态才具有治疗作用。药物制造、成型、包装等工艺过程中均牵涉到升温过程,因此药物在不同温度下的多晶态以及多晶态转变研究具有特别的重要性。
本文以常见的卡马西平为例,采用耐驰公司的差示扫描量热仪DSC详细探讨了药物的多晶态转变行为,并通过吉布斯自由能函数讨论,比较了基于实验现象的动力学表述和严格的热力学表述。
#p#
实验方法
样品名称:卡马西平(卡巴米嗪);
分子式:C15H12N2O;
仪 器:NETZSCH DSC;
测评依据:熔点、焓、玻璃化转变温度,与晶型I 、III晶型转变有关的摩尔热容及稳定性的改变。
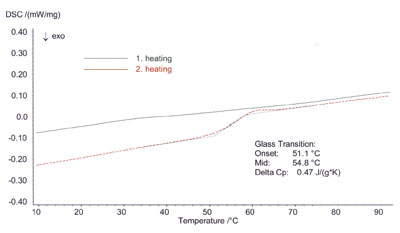
图2. 图1玻璃化转变部分放大图。
DSC测量结果
图1所示的卡马西平晶型III的第一次加热过程显示该物质在150~170℃的温度范围内有一个很宽的吸热过程,热值为2.5 KJ.mol-1(10.8 J.g-1)。样品在191℃出现熔融峰,相应熔融焓为23 KJ.mol-1(97 J.g-1)。将第一次扫描所得的熔融数据与文献值相比可得知样品物质在加热过程中发生晶型转变为晶型I。尽管熔融焓比文献值低很多,但熔点揭示了两种晶体形态上的差别。
#p#
这就产生了一个问题:为什么卡马西平的晶型III在160℃左右转变为晶型I?因为据文献报道,在176℃时晶型III才会熔解。以下是两个可能的原因:
(1)与晶型I相比,晶型III对热比较敏感,在所有温度下不稳定。
(2)晶型III在室温下是一个稳定的晶体,这是作为一个固态的市售药品的前提条件。
在第一次加热中, 会达到两种晶型稳定性相同的温度。当超过所谓的热力学转变点,晶型III就变得不稳定。根据热力学原理,晶型III将会转变为晶型I。两个可能的原因都可以解释图1中所观测到的现象。在热力学、动力学与晶体现象基础上,区分与阐明实验过程中的多相态关系, 如固-固、固-液、溶液-固态和固-气相态关系是一项非常困难的任务, 而且是不能简单解决的。要揭示卡马西平两种晶型的热力学状态, 吉布斯自由能函数或特殊的物理化学数据是必需的。这两项函数可由图5读出。
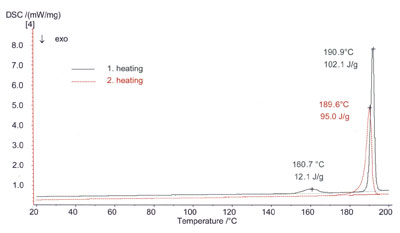
图3. 卡马西平,晶型III,连续熔融,1.06mg,封闭铝坩埚;升温:5~200℃/10K min-1;降温:200~ 5℃/10K min-1;升温:5~200℃/10K min-1。
在冷却过程中,可以从图1中清晰地看出另一种可能性:卡马西平以无定形形式出现。即在第二次加热中,玻璃化转变温度为55℃,热容变化值为δCp=0.47J.g-1.K-1。图1中玻璃化转变区域放大后显示为图2。无定形的卡马西平在受热时, 在106到121℃的温度范围里重结晶, 吸热为14 KJ.mol-1(61 J.g-1)。该样品在189℃出现熔融峰,显示为晶型I的熔点。熔融焓为20 KJ.mol-1(85 J.g-1)。与理论值相比,实验所测得的熔融焓值较低,即20 KJ.mol-1而不是26 KJ.mol-1,可解释为卡马西平的不完全重结晶或在一次加热中205℃时的分解。
#p#
另一个样品,晶型III在封闭坩埚中加热到200℃ (图3) 。其第一次加热的热力学行为和图1相同。熔点为191℃,熔融焓24 KJ.mol-1(102 J.g-1)。同时检测到液态在以10 K min-1的速率降温时,在172℃自发重结晶,结晶焓为22 KJ.mol-1(91 J.g-1)。第二次加热没有发现进一步的重结晶现象或固-固转变,仅在190℃出现晶型I的熔融峰, 熔融焓22 KJ.mol-1(95 J.g-1)。由于在第二次加热中发生玻璃化转变, 说明样品中含有少量无定形物质,而且也没有观测到再次结晶现象,这可以解释DSC曲线中较低的熔融焓值现象。根据熔点与熔融焓可得出以下结论:样品加热熔融后,在控制降温过程中,只有晶型I重结晶。
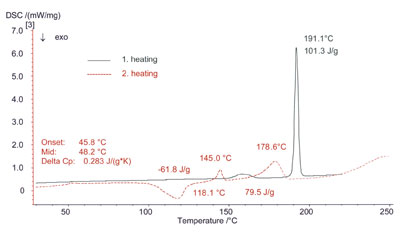
图4. 卡马西平, 晶型III, 连续熔融, 1.79mg, 铝坩埚, 带孔盖; 升温: 20~220℃/10K min-1; 降温: 220~ 20℃/10K min-1;升温: 20~250℃/10K min-1。
第三个样品第一次加热到220℃,得到了与前者相同的热力学行为。当以10K min-1的速率从220℃降温到20℃时,没有观测到重结晶现象。因此,可以推断无定形物质的存在。第二次加热过程中出现玻璃态转变肯定了这一点 (图4)。然后,出现了一个温度范围在90℃到130℃的放热重结晶过程。继续加热,在130℃至250℃观测到三个吸热效应。前两个在145℃与179℃的吸热峰可以解释为准共熔结晶二元混合物的熔融。样品在第一次加热后已经出现明显的分解, 在第二次加热中190℃以上出现的放热过程更清晰地表明了这一点。根据Griesser与Szelagiewicz等人的论文,卡马西平在165℃以上就已观测到分解现象。进一步的实验如TG-FTIR,TG-MS,X-ray光谱,IR和色谱分析等,对完全解释上述实验中的现象是十分必要的。
#p#
晶型转变研究
依据吉布斯自由能函数可以确定两种晶型的相互转变。晶形转变的关系有以下两种:单变性与互变性。在单变性系统中只可以观测到一个固态相转变,即所谓的由不稳定相态向稳定相态转变。互变性是一种完全不同的晶型转变关系。在互变性系统中存在一个热力学转变温度,它将稳定性分为两部分,仅在转变温度处,两种晶型的稳定性相同。
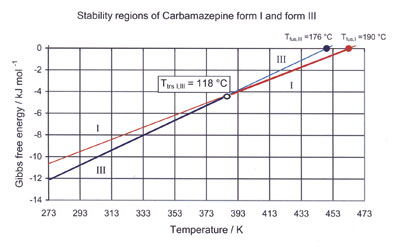
图5. 卡马西平晶型I与III转变的吉布斯自由能函数。
热力学转变温度可由吉布斯自由能函数的不同近似值导出。两种晶型的线性函数仅由熔点与熔融焓确定:
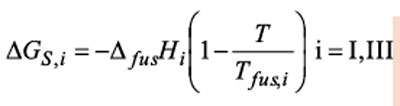
#p#
当满足下式时, 晶型I和III可能发生热力学转变:
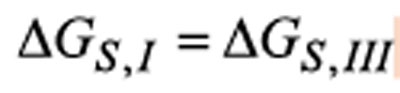
方程式 (1) 与 (2) 代入 (3) 可得出热力学转变温度:
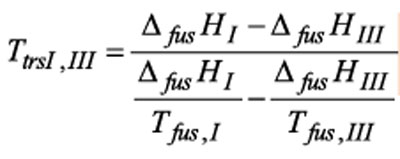
为了计算热力学转变温度,必须知道晶型熔点 (由绝对温度K表示) 和熔融焓。根据文献报道的晶型I、III热力学参数可以计算得到转变温度为TtrsI,III=118℃。
Heiber与Marti利用其实验数据,通过线性近似的自由能公式计算了晶型I、III的热力学转变温度。他们所得的数值是120℃。在30℃到150℃的温度范围内检测了两种晶型的比热容,结果显示两种函数关系没有显著的不同。在整个温度范围内的偏差小于0.003 J.g-1 K-1。因此热力学转变温度与近似级数没有显著联系。采用高价近似和实测的摩尔热容数值,得到的热力学转变温度为TtrsI,III=115±5℃。
#p#
如上面所提到的那样,在DSC曲线上160℃附近的固态转变 (如图1,3所示),可以解释为晶型III比晶型I不稳定造成的。然而,该转变过程可以在118℃以上的任一温度观测到。
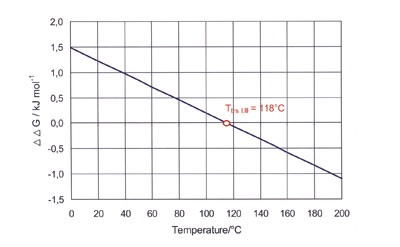
图6. 卡马西平,晶型I与III的δδG=δGs,I-δGs,III。
当然,在晶型III的熔点176℃,完全熔解的样品重结晶变为晶型I时,表现为放热反应,该过程的推动力是由δδG提供的 (图6)。晶型I与III之间转变的推动力可由下式计算:
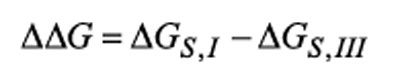
#p#
根据定义,在晶型III的熔点Tfus,III=176℃时,吉布斯自由能δGs,III为零,因此我们得到:
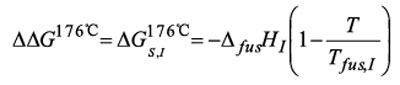
根据实验数据得到δδG176℃=-0.8 KJ.mol-1。
δδG的数值降低说明,在176℃晶型I的稳定性高于液相。多晶型转变,即熔点附近不稳定晶型的转变,不但是热力学决定的,而且液-固转变比固-固转变更有利。相对于固-固转变环境,液-固转变时不稳定晶型的熔融焓不予考虑,而且在液体状态下,分子的流动性要高几个数量级。
从118℃到晶型I的熔点,该温度范围内晶型III的转变是一个动力学过程,受许多因素影响。样品的化学纯度、物理纯度,即是否存在卡马西平的不同晶型,特别是是否存在晶型I作为晶体种子都是十分重要的。在另一方面,其它实验条件,例如样品大小、坩埚材质或加热速率都会影响转变的动力学。因此总结如下:在给定温度范围内的任何温度都可以观测到晶型III (或者液相) 转变到晶型I。
图4中所示的实验中,熔融的卡马西平冷却至室温,没有观测到重结晶现象。第二次加热中,样品在48℃时出现玻璃化转变,伴随的比热容变化δCP=0.28J.g-1K-1。玻璃化转变证明了在样品盘中出现了无定形态物质。在101℃至128℃中出现的再结晶主要转变为晶型I。对于140℃至179℃的典型二元体系共熔熔融,表明在第一次扫描到220℃时,样品发生了显著的分解。文献报道的卡马西平的显著不稳定性的起始温度都在165℃以上。
结论
第一次扫描与第二次扫描的DSC曲线可以表征晶型III的热力学行为,观测到固态相转变。要想深刻理解卡马西平多晶型的复杂性,需要在DSC测量中选用恰当的实验条件与合适的样品。DSC方法的使用已在实验部分的讨论中清晰阐明。
晶型转变的吉布斯自由能函数只能通过仔细地测量熔点与熔融焓,才能达到一定的准确度。另外,考虑到高活性晶型 (相对于稳定晶型而言) 的不稳定性,以及高熔点有机物的化学不稳定性,获得高准确度的热力学数据需要熟练的实验技术。
展源
何发
相关文章
-

QC, IQC, IPQC, QA,到底是什么鬼?
2020-05-27
-

AAS法分析茶叶中的铅,镉,砷
2020-05-27
-

检测有机氯类农药,气相色谱法检测法
2021-01-12
-

卡马西平的热分析(DSC)测量及转变热力学
2020-05-27
-
红外光谱分析,你了解多少?
2021-01-11
-

HPLC检测器,你了解吗?
2024-03-06
-

纯净的晶体--候选药物晶型的选择
2020-05-27
-

三聚氰胺,你还要害多少人
2020-05-27
-

药物研发晶型问题研究
2021-12-01
-

药物常用的晶型表征方法
2021-12-29

加载更多