诊断色谱峰裂分、前拖尾
色谱峰裂分、前拖尾的诊断
在液相色谱中分离中,几乎很少色谱图没有峰拖尾的情形。在反向色谱分离过程中,色谱峰拖尾主要是由于副反应造成的结果,尤其是在硅胶键合的固定相中碱性或酸性化合物与少量的金属杂质所发生的离子交换或相互作用。
这些峰拖尾的问题通常只是发生在色谱图中一个或少数几个色谱峰,但是对于那些所有色谱峰都出现峰拖尾、裂分或出现双峰的现象又该如何解释呢?另外有些色谱峰还会出现很糟糕的前沿,这对大多数色谱工作者来说并不多见,但在特定条件下却很普遍。本文将对这两种问题进行探讨。
反向使用色谱柱
曾有读者寄给我两张色谱图(图1),他在分析样品时发现,所有色谱峰均出现裂分,呈现双峰,见图1(a)。同一样品重复进样,结果依然如此。更换其它样品进样,同样色谱峰出现双峰。这时他觉得会不会所有的样品都会有同样问题。然后他用对照品重新进样,得到色谱图,见图1(b)。由于该实验室分析条件有限,只有一台LC,也没有多余的色谱柱,因此限制了他采用最有效的解决办法,用一个已知的好的配件来替代有问题的。尽管如此,他还是做他能做的,重新配制流动相及自动进样器清洗液,但是都没有得到改善。用乙腈充分冲洗整个色谱系统,使色谱柱、泵、自动进样器及检测器得到清洗,依然无法消除峰裂分的现象。最后,他无奈将色谱柱掉转方向使用,令人惊奇的是对照品的色谱峰由双峰变为了单峰。重新将样品溶液进样,同样,每一个组分均显示为单峰。由于担心色谱柱的安装方向与所指示的相反,他又将色谱柱重新按照通常的方向安装,结果以上所述问题再次出现,这就是他为什么写信求助的原因。
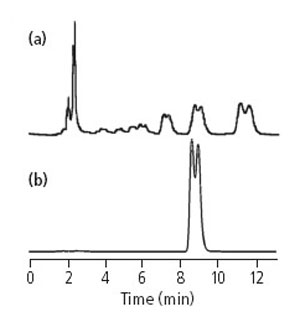
图1. 色谱柱部分堵塞导致的峰裂分色谱图,(a)为样品,(b)为对照品。
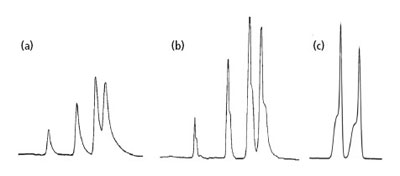
图2. 柱塞板部分堵塞(a, b)及柱塌陷(c)的色谱峰特征。
经典的色谱图
这个问题是很容易从色谱图看到的。事实上,这是一个很典型的我常常称之为“特征色谱图”的问题。它归结于两种色谱柱问题——柱筛板部分堵塞或者色谱柱头塌陷。图 2 所示即为这种问题导致色谱峰改变的例子。常见的现象为色谱图中每一个色谱峰均出现同样的变形,裂分成双峰,拖尾或其他意想不到的峰形。好的色谱柱情形如图3(a) 所示,色谱柱头的筛板使得样品均匀地分布在色谱柱的顶端。
由于这时还没有进行样品的分离,故每一股样品溶液都含有所有样品组分成分。它们在同一时间达到固定相,这时分离过程开始,在色谱柱中,化合物迁移速度的不同导致化合物的色谱分离。如果安装在色谱柱前端的柱筛板部分堵塞了,此时的情形如图3(b)所示,部分样品可以以通常的方式到达固定相表面,而另外一些则被堵塞的柱筛板挡住了,它们需要走更长的路绕过被堵塞的部分到达固定相,因此,这部分样品溶液就比其它没有被阻断的部分要晚些达到固定相。由于此时还没有进行柱分离,所以这个过程的结果导致一部分样品溶液先于其它部分开始流向色谱柱。就好象转眼之间发生了两次进样过程。从色谱图中就会看到双峰的出现,后者稍稍滞后前者。
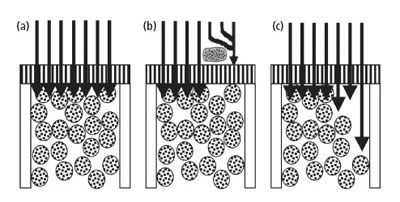
图3. 流动相通过色谱柱时的流向示意图,箭头所示为样品进入柱分离前段情形:(a)为正常流向,(b)为因柱筛板部分堵塞引起的流向改变,(c)为色谱柱塌陷引起的流向改变。
反向冲洗色谱柱或许能再次除去筛板上的污染物,因此可以恢复正常的流速及色谱峰形。有的时候反向冲洗可以解决峰裂分的问题,甚至将色谱柱再按正常方向使用。但在另外的情形中,反冲色谱柱可能是无效的或者只是在反向使用色谱柱时有效。可能是因为有些污染物强吸附在柱筛板上,在反向冲洗色谱柱时不能有效地将它们除去。
操作中的注意事项
反向冲洗色谱柱常常可以有效地除去吸附在色谱柱前端的颗粒污染物,但是需要提醒的是,第一,反向冲洗色谱柱时,要将出口端(原来的入口端)直接通入废液中几分钟,然后再接检测器,因为如果污染物被洗下来,你肯定不希望冲到检测器中,进而引起新的麻烦;第二,需要注意的是,色谱柱是否可以反向冲洗。如果色谱柱填料的粒径是5μm 或者更大,通常使用2μm 多孔柱筛板,此种色谱柱可以反向冲洗,不需要考虑太多,色谱柱上所表明的箭头流向只是一个参考的标识。如果色谱柱的填料是3μm,由于其分布有一较宽的粒径范围,2μm柱筛板其孔径通常不足够小,以便有效地挡住所有填料。因此,常常用0.5μm 柱筛板安装在色谱柱后端,0.5μm 孔径是与装在自动进样器后面用来保护色谱柱的在线过滤器的孔径一样的。这种筛板很容易被堵塞,同样的原因3μm填料的色谱柱比5μm的色谱柱更容易发生堵塞。消除柱堵塞的办法之一是色谱柱入口采用2μm的筛板,出口采用0.5μm的筛板。2μm的筛板不易被堵塞,而且只要按照色谱柱上所标识方向使用也可以保留住填料颗粒不流失。以我的经验,3μm填料配有2μm的筛板(入口)的色谱柱可以短时间反向冲洗,但不可长时间反向使用,否则可能会发生填料泄漏。对于最新的2μm填料的色谱柱,即使0.5μm的筛板,其孔径都太大了,故采用0.2μm的筛板。我不是很清楚厂家是否在色谱柱前段配有0.5μm的筛板,出口端配有0.2μm的筛板。但是即使是这种情况,色谱柱反向冲洗时仍要注意。在反向冲洗色谱柱之前,最好咨询色谱柱制造商有关色谱柱使用的注意事项,确保不损坏色谱柱。当然,最好的预防色谱柱堵塞的办法是尽量减少色谱柱被颗粒污染物污染,这意味着样品制备过程应包含除去杂质颗粒的过程。一些色谱工作者喜欢过滤所有样品溶液,这样做花费很高同时可能需要进行额外的验证来确保样品在过滤过程中不易丢失或者被过滤器带入污染物。我更建议采用台式离心机对所有样品离心,然后移入样品瓶中。
#p#
前沿峰
在反相色谱分离中,虽然峰拖尾现象无处不在,但前沿峰却不多见。一些色谱工作者可能从来没有见到过前沿峰。图 4 所示就是一个例子。该方法采用C18柱,流动相为含有高比例的水相(95%的缓冲液)和高pH值(pH 9.0)。在大约进样500次后,色谱柱出现异常,出现前沿峰。
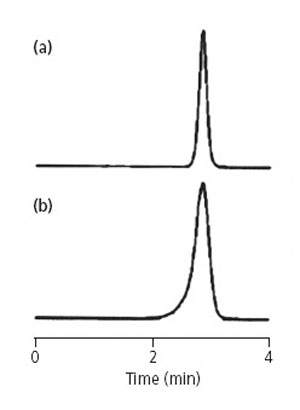
图4. (a)为正常色谱图;(b)柱为塌陷后的色谱图。
色谱峰拖尾的根源常被轻易地描述为样品(通常为碱性化合物)与硅胶键合柱的填充物发生化学副反应所引起的。但是前沿峰,更恰当的描述应该是色谱柱的物理变化造成的。色谱柱的塌陷导致样品溶液的流路发生改变,如图3(c)所示。在这个例子中,小部分样品溶液可以很快地流经塌陷色谱柱部分进入固定相表面,走在了其余样品溶液的前面,结果导致前沿峰的发生,如图4(b)所示。但样品中有多个色谱峰,则所有峰均显示峰前沿的现象,如图3(c)所示。在所列举的例子中,其色谱方法存在两个问题导致上述现象的发生。首先,流动相的pH值9.0,通常的高效液相色谱仪使用的硅胶基质色谱柱,其合适的pH值范围为2 < pH < 8。如pH值低于2,键合相被水解掉,如果pH值高于8,硅胶可以趋于溶解。有一些色谱柱,如使用复合硅胶键合相,可以使用pH高于8的流动相。另一种选择是用一种不易受到碱性物质腐蚀的聚合物填料的色谱柱。
另外的问题是图4 所示的方法需采用高含水量的流动相(95%缓冲液)进而使被分析物达到合适的保留。改善的方法之一是使用极性键合相或水型“AQ”色谱柱,此种色谱柱很多厂家都有制造,可以使用100%水相作为流动相;另外一种方法是减少硅胶表面C18的覆盖率,使更多的极性硅胶暴露在外面,至少有一家色谱柱制造商在使用该方法。色谱柱的类型如图4 所示。
不幸的是,高pH值的流动相加上低 C18覆盖率的色谱柱可能共同导致一些问题的发生,色谱柱在大约500次进样后塌陷,色谱峰出现很差的前沿峰,见图4(b)。幸运的是,色谱峰峰面积并没有受到影响,所以在该批样品定量分析尽管出现前沿峰,结果依然正常。当然,在接下来的样品运行之前,色谱柱被替换下来。能猜测到在方法的开发和验证过程中,这种致命的影响被忽视了,当意识到的时候,修正这一问题则需要进行方法的再验证。我们发现测试数据对于分析来说已经足够准确,并且分析方法运行超过10?000件的样品时也表现的不错。的确,更换色谱柱的花费很高,但是色谱柱的费用只是整个分析过程中的很小一部分开销。对于一个LC/MS/MS方法来说,一个样品的分析费用在在$50~$100,所以在连续进样500次以后,花$500来更换一个色谱柱,费用只占总费用的2%以下,远远低于上述例子中对方法进行再验证的花费。
结论
色谱柱进口端出现的物理问题可导致色谱峰裂分,双峰或者峰前沿。这些问题常常需要更换色谱柱来解决。如果色谱柱的寿命及数据质量都可以的话,不必改变方法来避免上述问题。
#p#
《实验与分析》
展源
何发
相关文章
-

色谱峰裂分,前拖尾的诊断
2020-05-27
-

色谱峰裂分、前拖尾的诊断
2020-05-27
-
色谱峰裂分、前拖尾情形的诊断
2020-05-27
-

色谱峰裂分,前拖尾的诊断
2020-05-27
-

色谱峰裂分和前拖尾的诊断
2020-05-27
-

气相色谱峰拖尾的3大原因
2022-07-07
-

解决峰拖尾与峰前延小妙招!
2023-06-30
-

气相色谱峰拖尾的原因及解决方法
2021-10-26
-

峰前延、分叉就是色谱柱坏了吗?
2023-12-14

加载更多